- Химические свойства спиртов
-
Химические свойства спиртов — это химические реакции спиртов во взаимодействии с другими веществами.
Они определяются в основном наличием гидроксильной группы и строением углеводородной цепи, а также их взаимным влиянием:
- Чем больше углеводородная цепь, тем сильнее она влияет на функциональную группу, снижая полярность связи O—Н. Реакции, основанные на разрыве этой связи, протекают медленнее.
- Гидроксильная группа −ОН уменьшает электронную плотность вдоль прилегающих σ-связей углеродной цепи (отрицательный индуктивный эффект).
Все химические реакции спиртов можно разделить на три условные группы, связанные с определёнными реакционными центрами и химическими связями:
- Разрыв связи O−H (реакционный центр — водород);
- Разрыв или присоединение по связи С−OH (реакционный центр — кислород);
- Разрыв связи −СOH (реакционный центр — углерод).
Реакции с участием гидроксильной группы (связи С−O и О−H)
Кислотно-основные реакции спиртов
Со щелочными и щелочноземельными металлами, алюминием, галлием, таллием и некоторыми другими металлами, а также сильными основаниями (например: амидами или гидридами) спирты способны реагировать с образованием алкоголятов[1]:
С сильными кислотами Льюиса спирты ведут себя подобно основаниям, образуя донорно-акцепторные комплексы[2]:
- Подробнее о природе кислотно-основных свойств спиртов: Диссоциация и кислотно-основные свойства спиртов.
Превращение спиртов в галогеналканы
Одной из наиболее важных реакций с участием связи C−O является превращение спиртов в галогеноалканы. Гидроксильная группа в спиртах может быть замещена на атом галогена несколькими способами[2]:
- взаимодействием с галогенводородами (HCl, HBr, HI);
- реакцией с тионилхлоридом;
- действием галогенидов фосфора (III) и (V);
- реакцией с квазифосфониевыми солями;
- превращением в алкилсульфонат с последующей реакцией замещения.
Взаимодействие спиртов с галогенводородами
Взаимодействие спиртов с галогенводородными кислотами приводит к замещению гидроксильной группы на галоген:
В зависимости от строения субстрата возможны побочные процессы изомеризации и дегидратации. Из-за относительно жестких условий проведения данные реакции применимы только к соединениям, устойчивым к кислотам.
Бромоводородную и иодоводородную кислоты часто получают непосредственно в ходе реакции из соответствующих солей (KBr, KI и т. д.) действием серной или фосфорной кислот[3][4].
Незамещенные первичные спирты превращаются в алкилбромиды с помощью горячей концентрированной бромоводородной кислоты[4]:
Попытки получить алкилиодид с помощью HI иногда могут приводить к восстановлению первоначального продукта до алкана. Помимо этого, свободный иодоводород способен реагировать с серной кислотой, приводя к образованию сернистой кислоты и иода[3]. Если субстрат содержит двойные связи, последние также могут быть восстановлены[5].
Вышеуказанные реакции можно использовать для получения первичных, вторичных и третичных галогеноалканов, хотя в случае изобутилового и неопентилового спиртов велики выходы продуктов перегруппировки[4][6].
Реакции третичных спиртов с HCl протекают достаточно легко. При этом образуются соответствующие третичные алкилхлориды (совместно с продуктами побочных реакций). Первичные и вторичные спирты реагируют гораздо медленнее и требуют применения катализатора. Обычно используется так называемый реагент Лукаса, представляющий собой смесь HCl и ZnCl2[4][7].
Хорошие выходы первичных алкилхлоридов были также получены при использовании HCl в HMPA (гексаметилфосфотриамид, биполярный апротонный растворитель)[8].
Прямое взаимодействие спиртов с фтороводородом возможно только при использовании третичных, аллиловых и бензиловых спиртов. Так, например, реакция трет-бутилового спирта c 60 % водным раствором HF при нагревании приводит к образованию трет-бутилфторида[9]:
Вместо чистой HF для фторирования обычно используют 70 % раствор фтороводорода в пиридине, так называемый реактив Олаха.
Первичные и вторичные спирты реагируют с галогенводородами по механизму SN2 (общая схема):
Для третичных спиртов характерен механизм SN1:
В ходе такого замещения образуется промежуточный карбокатион, поэтому SN1 реакции могут сопровождаться перегруппировками и элиминированием. Таким образом, практический интерес представляют только те третичные спирты, которые дают карбокатион, не способный к перегруппировкам.
Взаимодействие спиртов с галогенидами фосфора
Распространённым способом превращения спиртов в алкилгалогениды является их взаимодействие с галогенидами фосфора: нуклеофильному механизму с образованием галогенфосфита в качестве интермедиата[10]:[стр. 142—143]:
Для повышения выхода конечного продукта и уменьшения доли побочных реакций замещение ведут в присутствии пиридина.
В соответствии с особенностями механизма реакции (SN2), замещение гидроксильной группы на галоген происходит с обращением конфигурации у асимметрического атома углерода. При этом следует учитывать, что замещение часто осложняется изомеризацией и перегруппировками, поэтому подобная реакция, обычно, применяется для относительно спиртов простого строения[10]:[стр. 142].
Взаимодействие спиртов с тионилхлоридом
В зависимости от условий взаимодействие спиртов с SOCl2 протекает либо по механизму SNi, либо по механизму SN2. В обоих случаях спирт превращается в соответствующий алкилхлорид.
Если реакция проходит в отсутствие пиридина, продукт имеет такую же конфигурацию реакционного центра, что и исходный спирт (механизм SNi):
Добавление пиридина в реакционную смесь приводит к изменению стереохимического результата процесса. Полученный алкилхлорид имеет обращенную конфигурацию. Этот факт можно объяснить следующим механизмом SN2[4]:
Взаимодействие спиртов с хлорангидридами сульфокислот и последующим замещением
Спирты способны реагировать с хлорангидридами сульфокислот в присутствии основания с образованием соответствующих сложных эфиров. Первичные спирты реагируют быстрее вторичных и значительно быстрее третичных[4]. Возможно селективное образование первичного сложного эфира сульфокислоты в присутствии вторичных и третичных спиртовых групп. Наибольшее практическое значение имеет получение алкилтозилатов (R−O−SO2C6H4CH3), алкилмезилатов (R−O−SO2CH3) и алкилтрифлатов (R−O−SO2CF3). В роли основания чаще всего используется пиридин, который одновременно выступает и как нуклеофильный катализатор[4].
Сульфонаты являются прекрасными уходящими группами и легко замещаются на атом галогена по механизму SN2:
Источником галогенид-иона обычно является соответствующая неорганическая соль (NaBr, LiCl, CsF, KF] и т. д.) В качестве растворителя используются диполярные апротонные растворители: ДМСО, ДМФА, ацетонитрил. Замещение происходит, как правило, с обращением конфигурации[11]:[стр. 9].
Метод замещения гидроксила на высокореакционноспособную группу — мощный препаративный метод в органической химии, позволяющий получать из спиртов в две стадии, помимо галогенидов, самые различные соединения: простые эфиры, сложные эфиры карбоновых кислот, амиды и пр[10]:[стр. 151—152].
Взаимодействие спиртов с квазифосфониевыми солями
Спирты могут быть превращены в алкилгалогениды реакцией с квазифосфониевыми солями — [R3PHal]+X−. Последние образуются при взаимодействии органофосфионов (R3P) с галогенами, тетрагалогенметанами (CCl4, NBS). Данный метод применим к первичным и вторичным спиртам; в случае третичных спиртов возможно образование продуктов перегруппировки[2]. R3PBr2 и R3PI2 (получаются из R3P и Br2/I2) дают хорошие выходы даже с третичными и неопентильными субстратами[4]. В общем виде реакция протекает по следующей схеме[12]:
Превращение происходит с инверсией реакционного атома углерода[12].
Частный случай взаимодействия — превращение спиртов в алкилхлориды под действием трифенилфосфина и тетрахлорметана — в заграничной литературе получил название реакции Аппеля (англ. Appel reaction)[13][14]:
Прочие методы замещения гидроксильной группы на галоген
Приведём примеры некоторых дополнительных агентов, позволяющих провести замещение гидроксильной группы на галоген.
- Замещение OH− на F−:
- Одним из наиболее известных прямых фторирующих агентов для для первичных и вторичных спиртов является реагент Яровенко или N,N-диэтил(2-хлор-1,1,2-трифторэтил)-амин[15]:[стр. 87]:
- Удобным фторирующим агентом для первичных и вторичных спиртов может служить тетрафторид серы SF4[9]:
- По аналогии с SF4, можно использовать и [16].
- Среди современных фторирующих агентов для спиртов используют N,N-диэтиламиносеры трифторид (C2H5)2N-SF3 (англ. DAST), бис(2−метоксиэтил)аминосеры трифторид (CH3OC2H4)2N-SF3 (англ. Deoxofluor) и ряд других[17].
- Удобным методом конверсии спиртов в алкилфториды с выходом, близким к количественному, является их синтез через фтороформиаты (реакция с COF2, образующимся in situ из бис-(трихлорметил)карбоната и KF) с последующим разложением образующихся полупродуктов при 120—125 °С в присутствии гексабутилгуанидин фторида (HBGF) как катализатора[18]:
- Замещение OH− на Cl− и Br−:
- Эффективным методом замещения гидроксильной группы на Cl− и Br− является реакция спиртов при комнатной температуре с цианурхлоридом (цианурбромидом) и N,N-диметилформамидом в среде метиленхлорида[19]:
- Другим вариантом замещения гидроксильной группы на галоген является использование в качестве нуклеофильных агентов триметилсилилгалогенидов. При этом возможно замещение на I, Br и Cl — для последнего в качестве катализатора используются небольшие количества диметилсульфоксида[К 1][20]:
- Замещение OH− на I−:
- Удобным препаративным методом замещения гидроксильной группы практически всего диапазона спиртов (включая третичные, аллильные и бензиловые спирты) на иод, является использование в качестве регента соли трифенилфосфита с метилиодидом[11]:[стр. 8].:
- Новым методом получения алкилиодидов из первичных и вторичных спиртов является использование в качестве иодирующего агента тиоиминиевой соли в присутствии имидазола[21]:
Превращение спиртов в эфиры неорганических кислот
Получение нитратов и нитритов
Этерификацией спиртов концентрированной азотной кислотой получают органические нитраты[22]:
Используя нитрозирующие агенты (NaNO2+H2SO4; NOCl; NOBF4 и пр.) по аналогии можно получить эфиры азотистой кислоты[23]:
Хорошим нитрозирующим агентом для спиртов также является раствор в ацетонитриле нитрита тетрабутиламмония (C4H9)4NNO2 в смеси с 2,3-дихлор-5,6-дицианобензохиноном и трифенилфосфином[24].
Получение сульфитов и сульфатов
Серная кислота способна давать при взаимодействии со спиртами при низких температурах кислые и средние эфиры (алкилсульфаты):
В лаборатории данный способ можно использовать только для низших спиртов (метанол и этанол), так как в прочих случаях велика доля продуктов дегидратации: алкенов и простых эфиров[25]:[стр. 22].
Помимо серной кислоты для синтеза алкилсульфатов используют оксид серы(VI), хлорсульфоновую или аминосульфоновую кислоту[26].
Взаимодействием спиртов с тионилхлоридом или диоксидом серы (в присутствии иода или брома) в пиридине можно получить органические сульфиты[27]:
Получение гипогалогенитов
Стандартным способом получения органических гипохлоритов из спиртов является действие на последние раствора гипохлорита натрия при охлаждении и отсутствии прямого солнечного света[15]:[стр. 62—63]:
Похожим способом можно получить из первичных спиртов гипобромиты, при этом для этанола наблюдается очень высокий выход (92 %)[28]:
Отметим, что в этанол при взаимодействии с гипогалогенитами в других условиях окисляется с образованием хлороформа, бромоформа или иодоформа (галоформная реакция)[29]:
Получение прочих эфиров неорганических кислот
Реакцией спиртов с некоторыми неорганическими кислотами, их ангидридами или галогенангидридами можно получить различные эфиры:
- фосфаты органические[30]:
- фосфиты органические[31]:
- бораты органические[32]:
- тиоцианаты органические[33]:
Превращение спиртов в простые эфиры
Взаимодействие алкоголятов с алкилгалогенидами или алкилсульфонатами (Реакция Вильямсона)
В отличие от спиртов, являющихся слабыми нуклеофилами, алкоголяты, образующие алкоксид-ионы RO− — сильные нуклеофилы и легко реагируют с алкилгалогенидами по механизму SN2 с образованием простых эфиров[34]:
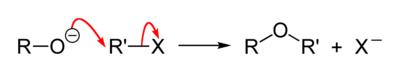
Вместо алкилгалогенидов можно использовать также алкилсульфонаты[25]:[стр. 21].
Побочными продуктами реакции являются алкены, образующиеся в результате конкурирующего процесса элиминирования спирта[34]:
Метод является одним из старейших в лабораторной практике и используется, преимущественно, для синтеза несимметричных эфиров[34]. Другим направлением использования реакции Вильямсона является синтез краун-эфиров[35].
Межмолекулярная и внутримолекулярная дегидратация спиртов
При осторожном нагревании в присутствии серной кислоты происходит межмолекулярная дегидратация спиртов с образованием простых эфиров[36]:
Если в реакцию с кислотой вступают двухатомные спирты, будет протекать реакция внутримолекулярной дегидратации с образованием гетероциклических соединений. Так например, 1,4-бутандиол образует тетрагидрофуран[36]:
Так как реакция получения эфира обратима, для её смещения вправо обычно используют метод отгонки конечных продуктов (воды или эфира) из реакционной смеси[36].
Существуют и методы термокаталитической дегидратации спиртов. Например, первичные спирты в присутствии смешанного Ni−Al2O3−SiO2 катализатора и водорода при нагревании превращаются в простые эфиры[37]:
Метод межмолекулярной дегидратации — один из наиболее старых способов получения эфиров — используется весьма ограниченно и только для неразветвлённых первичных спиртов из-за высокой доли алкенов, образующихся в случае внутримолекулярной дегидратации при использовании вторичных и третичных спиртов. Вместе с тем, реакция применяется в промышленности для синтеза некоторых простых эфиров[35].
Прочие методы превращения спиртов в простые эфиры
Среди прочих методов превращения спиртов в простые эфиры[38]:
- взаимодействие спиртов с диазометаном в присутствии кислот Льюиса;
- присоединение алкенов к спиртам в присутствии неорганических кислот.
Превращение спиртов в сложные эфиры
Кислотно-каталитическая реакция этерификации
Спирты способны образовывать сложные эфиры в реакциях с органическими кислотами при нагревании в присутствии кислотного катализатора (как правило, концентрированной H2SO4). Этот процесс получил название кислотно-каталитической реакции этерификации (также известен как реакция Фишера). Например, взаимодействие этанола с уксусной кислотой дает этилацетат[3]:
Механизм реакции[39]:
 Аппарат Дина — Старка в работе
Аппарат Дина — Старка в работе
Кислотно-каталитическая реакция этерификации — простейший и наиболее удобный метод получения сложных эфиров для случая, когда ни кислота, ни спирт не содержат чувствительных функциональных групп. В качестве катализатора, помимо традиционно используемой серной кислоты, могут выступать кислота Льюиса или Бренстеда; растворителем, обычно, служит сам спирт или, если это невозможно — толуол или ксилол. Для увеличения выхода эфира используют отгонку или химическое связывание воды, а также специализированное лабораторное оборудование — аппарат Дина — Старка[40].
Для пространственно затруднённых и склонных к элиминированию под действием кислот реагентов, например — трет-бутанола, существует метод мягкой этерификации, носящий имя этерификации Стеглиха (англ. Steglich Esterification). Реакция между спиртом и кислотой происходит в присутствии дициклогексилкарбодиимида (ДЦК) и небольших количеств 4-N,N-диметиламинопирнидина. ДЦК и карбоновая кислота на первом этапе образует O-ацилизомочевинный интермедиат, который в дальнейшем вступает в реакцию со спиртом, образуя сложный эфир[41]:
Реакция переэтерификации
Реакция переэтерификации или алкоголиза сложных эфиров имеет следующий общий вид:
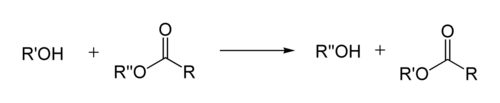
Для успешного осуществления переэтерификации используются различные методы: отгонка низкокипящих продуктов, использование специальных катализаторов, в том числе межфазного переноса и др. Механизм реакции переэтерификации аналогичен механизму гидролизау эфиров, поэтому в качестве побочного процесса возможно алкильное расщепление[42]:[стр. 130—131]:
Алкоголиз растительных жиров, представляющих собой сложные эфиры жирных кислот и глицерина, метиловым или этиловым спиртом — перспективная альтернатива производства биодизеля[43][44].
Взаимодействие спиртов с хлорангидридами, ангидридами кислот и нитрилами
С хлорангидридами карбоновых кислот спирты довольно легко вступают в реакцию, образуя сложные эфиры (реакция ацилирования)[25]:[стр. 20]:
Реакция спиртов с галогенангидридами — лучший общий способ получения сложных эфиров, так как позволяет использовать субстраты с самыми разными функциональными группами[42]:[стр. 125].
В 1898 году немецким химиком Айнхорном была предложена модификация этого метода: ацилирование проводится в избытке пиридина. Амин на первом этапе реагирует с хлорангидридом с образованием пиридиниевой соли, которая благодаря высокой ацилирующей способности под действием спирта легко трансформируется в эфир[45]:
Вместо ацилгалогенидов, для синтеза сложных эфиров может быть использована схожая реакция ангидридов карбоновых кислот со спиртами. В качестве катализаторов используют кислоты, кислоты и основания Льюиса, а также — пиридин и N-4,4-диметиламинопиридин[42]:[стр. 126]:
Другой путь получения эфиров: взаимодействие спиртов с нитрилами кислот в среде безводного хлороформа в присутствии газообразного хлороводорода приводит к иминоэфиру (реакция Пиннера), гидролизом которого можно получить сложный эфир[46]:
Этерификация Мукаямы
В 1975 году Мукаяма с сотрудниками предложил для достижения высоких выходов в реакции этерификации использовать специальный реагент — 2-хлор-1-метилпиридиния иодид[47]:
Метод Мукаямы в настоящий применяется для синтеза аминокислот и пептидов[48].
Реакция Мицунобу
При взаимодействии спиртов с карбоновыми кислотами в присутствии трифенилфосфина и диэтилазодикарбоксилата (англ. diethylazodicarboxylate, DEAD) образуется соответствующий сложный эфир. Данный процесс получил название реакции Мицунобу. Ключевой особенностью реакции является инверсия (обращение конфигурации) атома углерода при гидроксильной группе.
Механизм реакции Мицунобу[49][50][51]:
Прочие реакции замещения гидроксильной группы
Хлорокарбонилирование спиртов
Фосген COCl2 — источник хлоркарбонильной группы −C(O)Cl — способен реагировать с различными нуклеофильными агентами и, в частности, со спиртами в соответствии с механизмом SN1 или ацильным механизмом замещения (тетраэдрический переходный интермедиат)[52]:[стр. 46]:
В случае рассматриваемой реакции: R=L=Cl, Nu=RO−.
Алифатические спирты легко вступают в реакцию с фосгеном при комнатной температуре, образуя хлорформиаты (ROC(O)Cl) с высоким выходом[52]:[стр. 47]. Побочным продуктом реакции являются алкилхлориды, образующиеся при разложении хлорформиатов[52]:[стр. 49]:
Аналогично фосгену, в реакцию хлорокарбонилирования со спиртами вступают и его производные: дифосген, трифосген, оксалилхлорид.
Замещение гидроксильной группы на амидную
Нуклеофильное замещение гидроксильной группы на амидную возможно только в случае её модификации: перевода в оксониевую форму (−O+H2) под действием сильных кислот или предварительно получая диалкиловые эфиры серной кислоты (R−OSO2O−R), с последующим их замещением:
Прямое каталитическое взаимодействие простейших спиртов с аммиаком имеет исключительно промышленное значение, так как в его результате образуется смесь продуктов[53]:[стр. 517].:
Например, взаимодействием амилового спирта с аммиаком в присутствии водорода и катализаторов (Ni+Cr2O3) при повышенной температуре и давлении получают смешанные амиламины[54].
Взаимодействие спиртов с аммиаком в присутствии катализаторов дегидрирования (медь, никель, кобальт на оксиде алюминия и пр.) осуществляется через механизм дегидрирования с последующим аминированием[55]:
Также в промышленности используют конденсацию некоторых аминов со спиртами. Например, анилин в жёстких условиях (170—280 °С, давление 10 МПа, катализаторы: минеральные кислоты, никель) реагирует со спиртами с образованием смеси моно- и дизамещённых производных[56]:
Третичные спирты, обладающие подвижной гидроксильной группой, реагируют с мочевиной, образуя N-алкилпроизводные[57]:[стр. 77]:
Замещение гидроксильной группы на меркаптогруппу
Замещение гидроксильной группы на меркаптогруппу (−SH) с образованием тиолов можно осуществить действием на спирты P4S10 или взаимодействием паров спирта с сероводородом в присутствии гетерогенного катализатора[58]:
Альтернативным методом получения органических производных сероводородной кислоты является взаимодействие спиртов с тиомочевиной в кислой среде с последующим гидролизом[59][57]:[стр. 72]:
Замещение гидроксильной группы на нитрильную
По аналогии с синтезом амидов, замещение гидроксильной группы на нитрильную производят через получение алкилсульфоната, который в дальнейшем при действии цианида натрия или калия приводит к нитрилу[60]:[стр. 434].:
Замещение гидроксильной группы на азидную
Спирты не вступают в прямое взаимодействие с неорганическими азидами, однако их нагревание с азидом дифенилфосфорила позволяет в одну стадию перейти к органическим азидам[61]:
Реакции элиминирования спиртов
Кислотно-каталитическая дегидратация
Кислотно-каталитическая дегидратация спиртов — один из наиболее простых и доступных методов получения алкенов; при этом в качестве дегидратирующего агента возможно использование различных минеральных и органических кислот (серная, фосфорная или щавелевая кислота), кислых солей (гидросульфат натрия), а также кислот Льюиса[15]:[стр. 90].
В ненуклеофильной среде спирты, подвергаясь протонированию со стороны кислоты, элиминируются по механизму Е1. Механизм E2 для реакции дегидратации встречается редко[62]:[стр. 260—261]:
Образующийся в процессе реакции карбкатион склонен к проявлению H+-сдвига (миграции протона или алкильных групп), что приводит к перегруппировкам и получению в ходе элиминирования смеси конечных продуктов[62]:[стр. 261—262]:
Расщепление спиртов практически во всех случаях происходит по правилу Зайцева, то есть атом водород элиминируется от наименее гидрогенизированного атома углерода.
Оригинальным методом дегидратации является обработка алкоголятов соответствующих спиртов бромоформом (элиминирование происходит через образование промежуточных карбониевых интермедиатов)[63]:
Главным недостаткам кислотно-каталитической дегидратации спиртов является ограниченная возможность контроля положения образующейся двойной связи, а также структуры углеводородной цепи, поэтому данный метод, как правило, используется для стерически симметричных спиртов или спиртов, имеющих простое строение[64]:[стр. 175—176].
Термокаталитическая дегидратация
Термокаталитическая дегидратация спиртов над металлоксидными катализаторами — другой распространённый способ лабораторного получения алкенов[65]. Существует множество различных катализаторов дегидратации среди которых: Al2O3[66], ThO2[67], ZnO[68], V2O5[69], оксиды редкоземельных металлов (Ho2O3, Er2O3, Tm2O3, Yb2O3, Lu2O3, Y2O3, CeO2)[70], цеолиты[71].
Реакция дегидратации осуществляется при высокой температуре, при этом в качестве побочных процессов наблюдается дегидрирование спиртов[65][68]:
Исследования показали, что продуктами термической дегидратации на алюмооксидном катализаторе являются термодинамически более стабильные транс-алкены[72].
Недостатком термической дегидратации спиртов, как и каталитической дегидратации, является отсутствие контроля положения двойной связи, а также невозможность использования этого метола для соединений, содержащих различные термически неустойчивые функциональные группы.
Дегидратация с использованием специальных агентов
Дегидратация по Бургессу
Термолиз вторичных и третичных спиртов с метил N-(триэтилдаммонийсульфонил)карбаматом (реактив Бургесса) мягко и селективно приводит к алкенам[73]:
Реактив Бургесса применяется в каталитических количествах, при этом реакция идёт стереоспецифично и представляет собой цис-дегидратацию[74]:[стр. 732].
Дегидратация по Мартину
Наряду с реактивом Бургесса, для дегидратации спиртов используется ещё один органический реагент: сульфуран Мартина или дифенилбис(1,1,1,3,3,3-гексафторо-2-фенил-2-пропокси)сульфуран[74]:[стр. 811]:
Этот дегидратирующий агент используют, преимущественно, для вторичных и третичных спиртов, при этом последние реагируют с ним практически мгновенно; первичные спирты нереакционноспособны — образуют с сульфураном простые эфиры[75].
Элиминирование по Чугаеву
Реакция Чугаева — взаимодействие спиртов с CS2 и NaOH с последующим пиролизом образующегося ксантата[76]:
Продуктом реакции являются алкены, как правило, в цис-конфигурации. Главное преимущество метода — сведением к минимуму изомеризации и миграции кратной связи.
Строго говоря, элиминирование по Чугаеву аналогично получению алкенов пиролизом различных сложных эфиров. Подробнее см. статью Алкены.
Реакции окисления спиртов
Первичные спирты в зависимости от выбора агента окисляются до альдегидов или карбоновых кислот, вторичные — до соответствующих кетонов и кислот. Третичные спирты устойчивы к окислению, однако под действием сильных окислителей могут быть расщеплены с разрывом углеродной цепи в различные карбонильные соединения.
Реакции окисления спиртов, иначе — превращения в карбонильные соединения, можно разделить на две условные группы[77]:[cтр. 114]:
- реакции химического окисления;
- реакции каталитического дегидрирования.
В таблице 12. приведены сводные данные по реакциям окисления спиртов до различных производных[77]:[cтр. 303—305].
[T 1]Таблица 12. Окисление первичных, вторичных и третичных спиртов до различных производных.
Исходное соединение Конечное соединение Окислитель Катализатор дегидрирования Первичные спирты R−CH2OH R−CHO AgO, N2O4, K2Cr2O7 + H2SO4, CrO3, Ag2Cr2O7, (C5H5NH)2Cr2O7, C5H5NHCrO3Cl, CrO2Cl2, [(CH3)3CO]2CrO4, MnO2, K2FeO4, NiO2 Специфические агенты для Ar−CH2OH: (NH4)2Ce(NO3)6, NaBrO3, Pb(CH3COO)4, KOCl, (CH3)3COCl
Cu, CuO, Co2O3, Cr2O3, Ag, Pt, PtO2 R−CH2OH R−COOH O2/PtO2, HNO3 (конц.), H2CrO4 + H+, KMnO4 + H+, NiO2, Na2RuO4 PtO2 Вторичные спирты R−CHOH−R R−CO−R (NH4)2Ce(NO3)6, K2Cr2O7 + H2SO4,CrO3, [(CH3)3CO]2CrO4, H2CrO4, (C5H5NH)2Cr2O7, C5H5NHCrO3Cl, Br2, Cl2, NaOCl, Ca(OCl)2, NaBrO2, NaBrO3, MnO2, KMnO4, Ba(MnO4)2, K2FeO4, RuO4, Na2RuO4 Cu, CuO, CuCr2O4, Ni Ренея, Ag, Pd, Pt, PtO2 Третичные спирты (R)3C−OH (R)3C−O−OH H2O2 + H2SO4 — (R)3C−OH R−CO−R Pb(CH3COO)4 — (R)3C−OH RCOOH + R−CO−R CrO3 — Окисление неорганическими окислителями
Окисление соединениями хрома
В лабораторной практике для окисления спиртов чаще всего пользуются шестивалентными соединениями хрома: дихроматом натрия с серной кислотой или оксидом хрома(VI)[78]:[стр. 436]:
Обычно, для проведения реакции используют так называемый реагент Джонса — раствор оксида хрома(VI) в разбавленной серной кислоте и ацетоне. Реагент также может быть получен из дихромата натрия или калия. Окисление по Джонсу применяют для селективного окисления вторичных спиртов до кетонов и первичных спиртов до карбоновых кислот и в некоторых случаях до альдегидов[79].
Третичные спирты под действием триоксида хрома окисляются с разрушением углеводородного скелета, например, циклоалканолы трансформируются с раскрытием кольца в кетоны и карбоновые кислоты[80].
Альтернативой реагенту Джонсу является комплекс триоксида хрома с пиридином CrO3•2C5H5N, носящий имя реагент Саррета. Этот реагент позволяет проводить селективное окисление самых различных первичных спиртов до альдегидов в неводных условиях, однако его высокая пожароопасность и гигроскопичность, а также основные свойства пиридина ограничивают возможности применения[81].
Раствор реагента Саррета в метиленхлориде называется реагентом Коллинза. Эта модификация окислителя является более удобной и безопасной, а также может быть использована (в отличие от двух предыдущих реагентов) для окисления субстратов, чувствительных к действию кислот или щелочей[82].
В 1975 году для окисления спиртов в карбонильные соединения был предложен новый стабильный и удобный реагент на основе шестивалентного хрома — хлорохоромат пиридиния C5H5NHCrO3Cl[83]:
Схематичный механизм реакции[84]:
Важным достоинством реагента является его инертность по отношению к ненасыщенным связям, что позволяет получать непредельные альдегиды и кетоны.
Среди других комплексных соединений хрома используются: дихромат пиридиния, фторохромат пиридиния, хлорохромат дипиридиния, а также хлорохроматы различных гетероциклических соединений — хинолина, пиразина, имидазола и др[85].
Окисление соединениями марганца
Для окисления спиртов из соединений марганца чаще всего используют MnO2 и KMnO4. Варьируя условия проведения реакции (температура, pH среды и пр.) продуктами окисления могут стать альдегиды, кетоны или карбоновые кислоты.
Непредельные спирты при действии оксида марганца(IV) при комнатной температуре в зависимости от строения превращаются в альдегиды или кетоны, сохраняя двойную связь[86]:
Аналогично реагируют и ацетиленовые спирты[86].
Важным фактором активности оксида марганца(IV) является метод его получения — лучшие результаты получаются при реакции перманганата калия с сульфатом марганца в слабощелочной среде[62]:[стр. 267].
Раствор перманганата в кислой среде действует как сильный окислитель, который превращает первичные алифатические спирты в карбоновые кислоты, а вторичные — в кетоны[87]:
В щелочной среде на холоду растворы перманганата со спиртами не реагируют[25]:[стр. 22].
Осторожное окисление бензилового спирта кристаллическим перманганатом калия в неводной среде в присутствии краун-эфира в качестве катализатора фазового перехода селективно приводит к бензальдегиду[88].
Каталитическое окисление кислородом
Окисление спиртов кислородом воздуха в присутствии катализаторов — распространённый способ получения карбонильных соединений (как правило — кетонов) в промышленности[89].
Одним из общих способов является использование в качестве катализатора порошкообразного серебра[90]:
Метанол окисляется кислородом воздуха до формальдегида в присутствии оксидов переходных металлов (например: Fe2O3) с выходом до 95 % (реакция Адкинса — Питерсона)[91]:
Каталитическое окисление этанола кислородом воздуха в присутствии оксида хрома(III) или оксида меди(II) — популярный демонстрационный опыт для учебных целей[92][93]:
Использование смешанного литий-серебро-алюминиевого катализатора даёт возможность осуществить прямое окисление этанола в окись этилена[94]:
Для окисления спиртов могут использоваться самые различные катализаторы, например оксид ванадия(V)[95], оксид рутения(IV)[96], ацетат палладия(II)[97] и ряд других.
Окисление прочими неорганическими окислителями
Существует большое количество неорганических соединений, которые могут быть использованы для окисления спиртов в те или иные производные. В таблице 13. приведены примеры использования некоторых реагентов.
[T 2]Таблица 13. Примеры неорганических реагентов, используемых для окисления спиртов.
Окислитель Исходное соединение Конечное соединение Условия реакции ацетат свинца(IV): Pb(CH3COO)4 Ar−CH2OH Ar−CHO раствор в пиридине, комнатная температура[98] R−CR'OH−CR'OH—R (RR')C=O уксуснокислый раствор, количественный выход[99] тетраоксид диазота: N2O4 R−CH2OH R−COOH хлороформ, 0 °С[100] гипохлориты: Ca(OCl)2, NaOCl, KOCl R−CH2OH / R−CHOH−R R−C(O)−OCH2R / R−CO−R уксусная кислота, 0 °С[101] нитрат диаммония-церия(IV): (NH4)2Ce(NO3)6 Ar−CH2OH Ar−CHO уксусная кислота, 50—100 °С[60]:[стр. 9]. феррат калия: K2FeO4 Ar−CH2OH + CH3OH Ar−COOCH3 дихлорметан, CuSO4, выход более 70 %[102] реагент Фетизона: Ag2CO3 / кизельгур R−CH(OH)−R / R−CH(OH)-CH2-CH(OH)−R R−C(O)−R / R−C(O)-CH2-CH(OH)−R карбонат серебра, нанесённый на твёрдый носитель кизельгур (англ. celite)[103] Окисление с использованием активированного диметилсульфоксида
Окисление Пфицнера — Моффатта
В 1963 году К. Пфицнером и Дж. Моффаттом была совершена публикация, в которой сообщалось об открытии нового метода окисления спиртов. Учёные растворяли исходные компоненты в смеси безводного диметилсульфоксида и дициклогексилкарбодиимида в присутствии слабой кислоты. В результате реакции в зависимости от строения спирта получался соответствующий альдегид или кетон, при этом даже для чувствительных первичных спиртов в продуктах окисления практически не наблюдались следов карбоновых кислот[104]:
Спустя два года был предложен механизм превращения[105][106]:
В соответствии с механизмом реакции протонированный дициклогексилкарбодиимид (ДЦК) на первом этапе вступает в реакцию с диметисульфоксидом (ДМСО) с образованием сульфониевого интермедиата (1), так называемого «активированного ДМСО», содержащего легко-уходящую группу, связанную с положительно заряженным атомом серы. Спирт быстро замещает эту группу, образуя алкоксидиметилсульфониевую соль (2), которая в свою очередь, теряя протон, превращается в тиоилид (3). В финальной стадии процесса происходит внутримолекулярное расщепление илида, проводящее к образованию конечного карбонильного соединения и диметилсульфида. Отмечается, что «Активированный ДМСО» (1) способен распадаться с образованием высокореакционной частицы (4), которая вступая в реакцию со спиртом, образует побочный продукт — метилтиометиловый эфир(5). Вместе с тем, учитывая, что элиминирование протекает при более высокой температуре, чем основной процесс, можно использовать температурный контроль хода реакции для минимизации доли побочных продуктов[105].
Согласно механизму окисления, для протонирования ДЦК необходимо присутствие кислоты, однако сильные минеральные кислоты (HCl, HClO4, H2SO4 и т. п.) для реакции непригодны — они предотвращают образование илида (3). Проведённые эксперименты показали, что оптимальным является использование фосфорной или дихлоруксусной кислоты, а также трифторацетата пиридиния[107].
Данный метод стал основой для многочисленных научных исследований в области окисления спиртов активированным диметилсульфоксидом, что привело впоследствии к многочисленным модификациям и практическим разработкам новых способов окисления[108]:[стр. 991—100].
Окисление Олбрайта — Голдмана и Олбрайта — Онодера
В 1965 году (спустя два года после сообщения Пфицнера и Моффатта) Олбрайтом и Голдманом был предложен способ окисления спиртов при комнатной температуре смесью ДМСО и уксусного ангидрида[106]. Предложенная модификация уступает методу Пфицнера — Моффатта из-за большего количества побочных продуктов, однако доступность уксусного ангидрида делает окисление Олбрайта — Голдмана полезным для лабораторной практики[108]:[стр. 114].
В том же сообщении 1965 года Олбрайт и Голдман упомянули, что ДМСО можно активировать оксидом фосфора(V)[106]. Спустя несколько месяцев Онодера с сотрудниками сделал подробный доклад о новом методе окисления спиртов смесью ДМСО и P2O5 (метод получил название окисление Олбрайта — Онодера[108]:[стр. 118])[109]. Наконец, в 1987 году данный способ окисления был улучшен: в качестве растворителя был использован дихлорметан в присутствии триэтиламина[110].
Окисление Париха — Деринга
Ещё одним методом окисления спиртов с использованием активированного диметилсульфоксида является окисление Париха — Деринга, где в качестве активирующего реагента используется раствор триоксида серы в пиридине (пиридиновый комплекс SO3•C5H5N) в присутствии триэтиламина. Реакция проходит при охлаждении (около 0 °С) или комнатной температуре. Метод, открытый Парихом и Дерингом в 1967 году, несмотря на его практическую доступность, отличается повышенным содержанием в целевых продуктах побочного компонента — метилтиометилового эфира. Механизм окисления Париха — Деринга аналогичен механизму окисления Пфицнера — Моффатта[111].
Окисление Сверна
Одним из лучших методов, использующих активированный ДМСО, стал процесс с использованием оксалилхлорида, открытый в 1978 году Сверном[112]:
Окисление спиртов по Сверну может быть выполнено в очень мягких условиях. С помощью этой реакции можно получать альдегиды и кетоны из первичных и вторичных спиртов соответственно. Главным недостатком метода является выделение токсичных и зловонных побочных продуктов — диметилсульфида и оксида углерода(II)[113].
Первый этап реакции Сверна заключается в низкотемпературном взаимодействии диметилсульфоксида (1a и 1b) с оксалихлоридом (2). Промежуточный интермедиат (3) быстро разлагается с выделением CO и CO2 и образованием хлорида диметилхлорсульфония (4), который в свою очередь вступает в реакцию со спиртом (5), образуя ион алкоксисульфония (6). Далее в реакцию вступает триэтиламин, который депротонирует интермедиат, давая илид (7). Переходный пятичленный цикл (7) разлагается, образуя диметилсульфид и конечный кетон или альдегид (8)[112][114].
Окисление Кори — Кима
В отличие от окисления по Пфицнеру — Моффатту и ему подобных, где «активированный ДМСО» образуется в реакции ДМСО с электрофильным агентом, метод Кори — Кима использует в качестве исходного реагента диметилсульфид[115]:
Сущность метода заключалась в образовании хлорида хлордиметилсульфония — представлявшего собой по сути «активированный ДМСО» Сверна (см. Окисление Сверна) — действием хлора на ДМС[116]:
На практике, однако учёные предложили использовать вместо хлора N-хлорсукцинимид (NCS), который вступая в реакцию с диметисульфидом, образует ион хлордиметилсульфония, а он в свою очередь реагирует со спиртом по аналогии с процессом Сверна[116]:
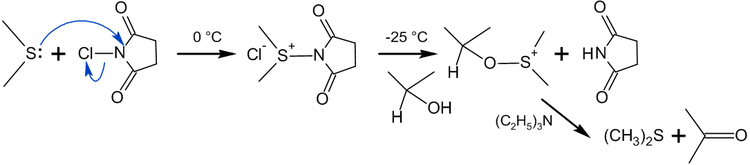
Окисление с использованием алкоголятов металлов
Окисление по Оппенауэру
В начале 20-го века независимо Меервейном, Пондорфом и Верлеем была открыта реакция восстановления карбонильных соединений в спирты (восстановление по Меервейну — Пондорфу — Верлею) в присутствии алкоголята алюминия (в качестве донора протонов выступал изопропанол)[117][118][119]:
В 1937 году Оппенауэром была осуществлена обратная реакция: используя в качестве окислителя избыток ацетона в присутствии трет-бутилата алюминия, ему удалось, по сути, сдвинуть равновесие и перенести процесс восстановления в обратную сторону[120][121]:
Окисление Мукаямы
В 1977 году Мукаяма с сотрудниками опубликовал работу, в которой сообщал, что алкоголяты магния, образующиеся в результате взаимодействия спирта с пропилмагнийбромидом или трет-бутоксимагнийбромидом в присутствии 1,1’-(азодикарбонил)дипиперидина (выступает в роли акцептора водорода) при комнатной температуре окисляют исходный спирт до альдегида или кетона[122]:
Хотя реакция Мукаямы и не принадлежит к числу распространённых методов окисления спиртов, она представляет препаративный интерес из-за более мягких условий протекания (по сравнению с окислением Оппенауэра) и сопровождается меньшим количеством побочных продуктов[108]:[стр. 276].
Прочие методы окисления
Окисление соединениями гипервалентного иода
Соединения пятивалентного иода — сильные окислители, однако из-за своей нестабильности и плохой растворимости в органических растворителях они практически не использовались в лабораторной органической практике. Однако, в 1983 году Десс и Мартин опубликовали информацию о новом стабильном и хорошо растворимом в дихлорметане органическом соединении гипервалентного иода, являющегося эффективным и очень мягким окислителем для первичных и вторичных спиртов[123].
Метод, получивший название окисления Десса — Мартина, оказался очень эффективным и получил свое развитие во многих последующих работах[124][125][126].
Помимо периодинана Десса — Мартина существуют и другие соединения гипервалентного иода, используемые как окислители для спиртов: 2-иодоксибензойная кислота, дихлорид иодбензола, иодозобензол и др.[126].
Окисление стабильными нитроксидными радикалами
 TEMPO — стабильный нитроксидный радикал
TEMPO — стабильный нитроксидный радикалВ 1987 году Анелли с сотрудниками опубликовал исследование, в котором сообщалось об использовании свободного нитроксидного радикала (4-метокси-2,2,6,6-тетраметилпиперидин-1-оксил или англ. 4-метокси-TEMPO) в качестве катализатора для быстрого селективного окисления первичных и вторичных спиртов. Реакция проводилась при 0 °С в двухфазной среде CH2Cl2—вода в присутствии вторичного окислителя (NaOCl), а также небольших количеств NaHCO3 (стабилизирует pH раствора) и KBr (ускоряет реакцию вследствие образования HOBr — более сильного окислителя по сравнению с HOCl)[127]:
Механизм реакции окисления с использованием TEMPO выглядит следующим образом[128]:
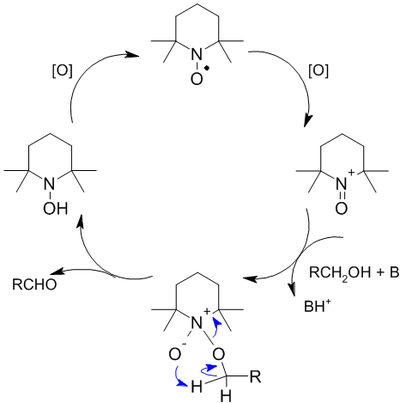
В настоящий момент окисление с использованием TEMPO и его производных широко распространено в лабораторной практике, при этом сама реакция неоднократно модифицировалась: в качестве вторичных окислителей помимо NaOCl используются иодозобензол, трихлороизоциануровая кислота, CuCl + O2, [129].
Каталитическое дегидрирование
Каталитическое дегидрирование спиртов — распространённый промышленный метод получения карбонильных соединений — в лабораторной практике используется редко из-за необходимости использования сложного оборудования и специально приготовленных катализаторов на основе меди, серебра, платины (в том числе с различными добавками), а также различных оксидов металлов, включая смешанные композиции[60]:[стр. 11—12].
Дегидрированием метанола над медью, хромом или серебром в промышленных масштабах получают формальдегид[130]:
При дегидрировании метанола на модифицированном медном катализаторе при температуре 200 °С образуется метилформиат[131]:[стр. 131]:
Высокодегидратированный оксид кремния(IV) может быть использован для селективного получения ацетальдегида из этанола[132]:
Одним из методов, используемых на практике довольно длительное время, является каталитическое дегидрогенирование первичных и вторичных спиртов в присутствии смешанного оксида меди-хрома при температуре около 300 °С[133]:
Современные препаративные методы дегидрирования спиртов на основе металлорганических катализаторов позволяют получать карбонильные соединения в мягких условиях и с высоким выходом. Например, сложный рутениевый комплекс можно использовать для трансформации первичных спиртов в кетоны[134] или сложные эфиры[135]:
Другой пример реакции с использованием комплексных органических соединений рутения в качестве катализатора[136]:
Биохимическое окисление спиртов
Особый способ окисления спиртов — биохимический, происходящий в живых организмах под действием естественных ферментов — является, с одной стороны, важным метаболическим процессом, с другой — промышленным микробиологическим процессом, используемым для получения различных полезных соединений.
Наибольшее практическое значение имеет способность ряда аэробных бактерий семейства Acetobacteraceae (роды Acetobacter и Gluconobacter) под действием кислорода в процессе клеточного дыхания трансформировать спирты в соответствующие карбонильные соединения или карбоновые кислоты. Наиболее важным из процессов подобного типа, является уксуснокислое брожение, общая схема которого выглядит следующим образом (для Acetobacter aceti):
В данной схеме катализаторами процесса являются следующие ферменты: алкогольдегидрогеназа (ADH) и альдегиддегидрогеназа (ALDH). Коферментом группы дегидрогеназ выступает пирохинолон хинон (PQQ)[137].
Известны и другие примеры биохимического окисления спиртов. Так, например, граммотрицательные бактерии Gluconobacter oxydans помимо уксуснокислого превращения этанола, могут трансформировать глицерин в дигидрокисацетон, маннит — во фруктозу, а сорбитол — в сорбозу[138].
Реакции восстановления спиртов
Каталитическое гидрирование
Неактивированные гидроксильные группы довольно устойчивы к гидрогенолизу и могут быть восстановлены в довольно жёстких условиях. Реакции гидрирования протекают при высоких температурах и давлении, в качестве катализаторов используются никель[139], смешанные оксиды хрома-меди[140], цеолиты[141].
В процессе гидрирования для высших спиртов параллельно может происходить укорочение углеводородной цепочки[142]:
Гидрирование первичных спиртов может быть описано как SN2 замещение с атакой водородом углеродного атома. Реакция третичных спиртов соответствует механизму SN1[141].
Гидрирование многоатомных спиртов может проходить с высокой степенью селективности. Так, например, гидрогенолиз глицерина можно остановить на стадии 1,2-пропандиола[142]:
Удобным методом гидрирования спиртов является двухстадийный процесс, на первом этапе которого под действием дициклогексилкарбодиимида в присутствии каталитических количеств CuCl спирт трансформируется в O-алкил-N,N-дициклогексилизомочевину, которая затем легко гидрируется в мягких условиях уголь-палладиевым катализатором[142]:
Реакция сочетания спиртов
Аллиловые и бензиловые спирты под действием системы метиллитий−хлорид титана (III) при −78 °С или при кипячении в присутствии алюмогидрида лития и хлорида титана (III) вступают в реакцию симметричного сочетания, согласно приведённой выше схеме. В случае использования смеси двух различных спиртов образуется соответствующая смесь трёх возможных продуктов сочетания[42]:[стр. 197].
Сочетание спиртов в присутствии рутениевого катализатора и кислот Льюиса происходит по другой схеме[143]:
Реакция Бартона — МакКомби
Одним из удобных и широко используемых методов восстановления спиртов до алканов является радикальное деоксигенирование тиокарбонатов и ксантогенатов в присутствии гидрида трибутилолова (или других источников водородных радикалов) и азобисизобутиронитрила (AIBN, инициатор радикального процесса)[144]:
Данный метод, получивший название реакции Бартона — МакКомби или реакции Бартона, имеет следующий механизм:
Восстановление спиртов другими методами
Одним из простейших способов восстановления спиртов является их взаимодействие с иодоводородом[145]:
На практике чаще пользуются смесью фосфора с иодом для замены дорогостоящего HI и регенерации иода в процессе реакции[145]:
Среди прочих восстановителей различных в литературе встречаются: иодистоводородная кислота в уксусном ангидриде, металлический цинк в комбинации с уксусной или соляной кислотами, натрий в жидком аммиаке и пр[53]:[стр. 14].
Реакции карбонилирования и гидроформилирования спиртов
Реакции карбонилирования спиртов
В 1953 году Реппе в своей работе показал, что в присутствии карбонилов кобальта, железа и никеля под действием высокой температуры и давления спирты способны присоединять оксид углерода (II) с образованием карбоновых кислот. Процесс получил название карбонилирование[146]:
Карбонилирование многоатомных спиртов приводит к поликарбоновым кислотам:
В дальнейшем процесс был усовершенствован: был применён кобальтовый катализатор с иодсодержащим промотором. Активным соединением в процессе является HCo(CO)4 (тетракарбонилгидрокобальт), образующийся в ходе реакций[131]:[стр. 134—135]:
Далее процесс идёт следующим образом:
Карбонилирование используется в промышленных синтезах и возможно не только для метанола: использование родия и других катализаторов позволяет присоединять CO к самым различным первичным, вторичным и даже третичным спиртам[131]:[стр. 137].
Реакции гидроформилирования спиртов
Для низших спиртов возможно осуществление и реакции гидроформилирования более характерной для алкенов[131]:[стр. 140]:
Более известна реакция гомологизации, то есть превращение органического соединения в свой гомолог путём внедрения одной или нескольких метиленовых групп, для спиртов была впервые осуществлена в 1940 году — на основе метанола каталитическим путём под воздействием высокого давления был синтезирован этанол[147]:
Гидроформилирование процесс крайне ограниченного использования — только немногие спирты (трет-бутанол, бензиловый спирт) дают приемлемые выходы и относительно высокую селективность[131]:[стр. 147].
Реакции окислительного карбонилирования спиртов
В 1963 году был впервые описан процесс окислительного карбонилирования спиртов в присутствии PdCl2 в качестве катализатора[148]:
Если реакцию проводить при повышенном давлении (7 МПа) и температуре (125 °С) конечным продуктом вместо диэтилкабоната будет диэтилоксалат.
Разработаны также схемы синтеза методом окислительного карбонилирования диметилкарбоната и диметилоксалата из метанола, дибутилоксалата из бутанола, а также ряда других соединений[148].
Прочие важные реакции с участием спиртов
Пинаколиновая перегруппировка
Дитретичные 1,2-диолы способны участвовать в реакциях пинаколиновой перегруппировки. В ходе процесса происходит 1,2-миграция алкильной группы в промежуточном карбкатионе. Продуктами являются пинаколины — кетоны, в которых карбонильнах группа соединена с третичным атомом углерода. Название реакции происходит от наиболее известного примера перегруппировки, превращения пинакола в пинаколон[149]:
Пинаколиновая перегруппировка относится к перегруппировке Вагнера — Mеервейна.
Перегруппировка Вагнера — Меервейна
При дегидратации спиртов алициклического ряда (содержащих насыщенный циклический фрагмент) возможно образование продуктов 1,2-миграции алкильной группы. Миграция направлена к карбкатионному центру в процессе элиминирования. Такие реакции, наряду с аналогичными в результате присоединения к кратным связям или нуклеофильного замещения, называются перегруппировками Вагнера — Меервейна. Особое значение реакция имеет для бициклических соединений, в частности — производных камфоры. Примером такой реакции может служить кислотно-катализируемое превращение изоборнеола в камфен[150].
Некоторые реакции присоединения с участием спиртов
Присоединение спиртов к соединениям, содержащим кратные связи, имеет важное значение в лабораторной практике.
Отметим кратко некоторые наиболее типичные реакции присоединения с участием спиртов.
- Реакция спиртов с альдегидами в присутствии катализаторов (HCl, n-толуолсульфокислота и др.) приводит к образованию ацеталей[29]:
- Спирты довольно легко присоединяются к эпоксидам, в результате чего образуются эфиры этиленгликоля (целлозольвы):
- Нагревание спиртов с нитрилами в присутствии газообразного HCl приводит к образованию иминоэфира (подробнее см. раздел: Взаимодействие спиртов с хлорангидридами, ангидридами кислот и нитрилами) :
- Аналогично реагируют и изотиоцианаты.
- При взаимодействии спиртов с сероуглеродом в щелочной среде образуются дитиокарбонаты (см. также раздел: Элиминирование по Чугаеву)[57]:[стр. 159]:
- Алкены в присутствии PdCl2 присоединяются к спиртам с образованием кеталей как основных продуктов[151]:
- Алкины присоединяют спирты в присутствии щелочей с образованием алкенильных эфиров (Реакция Фаворского):
Защита гидроксильной группы при органическом синтезе
Спирты, как правило, достаточно легко вступают в реакции нуклеофильного замещения с различными субстратами, способны окисляться до карбонильных соединений или терять воду под действием кислот. При проведении комплексных синтезов, часто появляется необходимость защиты гидроксильных групп для осуществления реакций в отношении других реакционных центров. Во время синтеза защищенная гидрокисльная группа остается без изменения, а по окончании процесса защита снимается с помощью специальных реагентов[152].
[T 3]Таблица 14. Некоторые распространённые защитные группы для спиртов, а также реагенты для их установки и удаления[153].
Защитная группа Установка защитной группы Снятие защитной группы реагент для установки среда для установки реагент для снятия среда для снятия CH3CO− (CH3O)2SO2 NaOH, (C4H9)4N+I− (CH3)3SiI CHCl3 (CH3O)2SO2 или CH3I NaH или KH, ТГФ BBr3 NaI, краун-эфир или CH3COOC2H5 CH3I KOH, ДМСО BF3•(C2H5)2O HCl (CH3)3COK, ТГФ SiCl4 NaI, CH2Cl2, CH3CN Ag2O AlCl3 или AlBr3 C2H5SH CH2N2 силикагель AlCl3 (C4H9)4N+I−, CH3CN (CH3)3CO− CH2=C(CH3)2 H2SO4 или H3PO4, BF3•(C2H5)2O HCl диоксан (CH3)3COС(=NH)CCl3 BF3•(C2H5)2O, CH2Cl2, циклогексан HBr CH3COOH (CH3)3SiI CHCl3 или CCl4 CH2=CHCH2O− CH2=CHCH2Br NaOH или NaH, бензол a. (CH3)3COK; b. H+ ДМСО (C2H5O)2Mg a. [(C6H5)3P]3RhCl, C2H5OH; b. Hg2+, H+ DABCO BaO, ДМФ PdCl2, CuCl, O2 ДМФ KF—Al, CH3CN NaOH Pd/H2 C2H5OH NaH, ТГФ, (C4H9)4N+I− Na в жидком аммиаке — Ag2O, ДМФ SnCl4 CH2Cl2 HCOOH (C2H5)2O Pd/H2 C2H5OH BF3•(C2H5)2O CH2Cl2, CH3OH (CH3)3SiO− лимонная кислота CH3OH CH3C(OSi(CH3)3)=NSi(CH3)3 ДМФ FeCl3 CH3CN CH3CH=C(OCH3)OSi(CH3)3 CH2Cl2 или CH3CN 2,3-дихлоро-5,6-дициано-1,4-бензохинон CH3COOC2H5 (C2H5)3SiO− пиридин HF CH3CN CsF, имидазол 2,3-дихлоро-5,6-дициано-1,4-бензохинон CH3CN HCOO− HCOOH — KHCO3 CH3OH СH3COO− (СH3CO)2O пиридин K2CO3 CH3OH СH3COCl CH2Cl2 гуанидин C2H5OH, CH2Cl2 СCl3COO− пиридин, ДМФ NH3 C2H5OH, CHCl3 СF3COO− пиридин H2О — С6H5COO− пиридин NaOH CH3OH бутиллитий (C2H5)3N CH3OH править] Защита через силильные эфиры Одним из наиболее распространённых способов защиты гидроксильных групп является реакция спиртов с хлортриалкилсиланом в присутствии основания с образованием алкилсилильных эфиров[154][стр.626—628]:
Образующийся силильный эфир можно далее использовать для сторонних синтезов, например:
Для снятия защиты используется кислотный гидролиз:
Защита через простые эфиры
Важным способом защиты гидроксильных групп является метод образования простых эфиров. В качестве таких соединений чаще всего используются трет-бутиловый, аллиловый, бензиловый, трифенилметиловый эфиры; реже — метиловый эфир[152].
Установка защиты происходит следующим образом[152]:
- трет-бутиловый эфир:
- аллиловый эфир:
- бензиловый эфир:
- трифенилметиловый эфир:
- метиловый эфир:
Для снятия защиты чаще всего используют кислотный гидролиз простых эфиров действием HI, HBr, ДМСО, являющегося сильным основанием («супероснованием»). Этот раствор способен генерировать карбанионные интермедиаты, которые достаточно легко вступают в реакцию с простыми эфирами[152]:
Защита через сложные эфиры
Защита гидроксильной группы возможна через образование сложных эфиров. Чаще всего для этих целей используются ацетаты, образующиеся при взаимодействии спиртов с уксусным ангидридом при комнатной температуре в пиридиновой среде; иногда в сочетании с некоторыми кислотными катализаторами[155]:[стр.110]:
Снятие защитной группы осуществляется с помощью основного (реже — кислотного) гидролиза, например: аммонолиза аммиаком в среде метанола[155]:[стр.111].
Для защиты гидроксильной группы в сахарах, помимо ацетатной, используется бензоильная и нитробензоильная защита (реагент — хлористый бензоил или нитробензоил). В химии стероидов находит применение формиатная защита (реагент — муравьиная кислота), которая избирательно (без затрагивания прочих сложноэфирных групп) может быть удалена гидрокарбонатом калия в метанольном растворе. Среди прочих защитных сложноэфирных групп отметим трифтор-, и хлор-, метокси- и феноксиацетаты, а также карбонаты и некоторые другие производные[155]:[стр.111—115].
Защита через ацетали и кетали
Одним из наиболее общих и эффективных методов защиты гидроксильных групп является количественная реакция спиртов с 2,3-дигидро-4H-пираном в условиях кислотного катализа (POCl3, HCl и др.). Для снятия защиты образующиеся тетрагидропираниловые эфиры могут быть подвергнуты кислотному гидролизу в достаточно мягких условиях[155]:[стр.104—107]:
Тетрагидропиранильная защита достаточна распространена из-за лёгкости установки и удаления, однако неприменима в условиях кислой среды и для оптически активных спиртов[155]:[стр. 104—107]. Если есть необходимость защиты стереоизомерных спиртов, для защиты используются симметричные ацетали или кетали и, в частности, метоксипроизводные дигидропирана[155]:[стр. 108—109].
Список таблиц
Комментарии
- ↑ При проведении реакции в других условиях образуются триметилсилильные эфиры, являющиеся хорошими защитными группами для спиртов. Подробнее см. раздел Защита через силильные эфиры.
Примечания
- ↑ Алкоголяты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 166—170.
- ↑ 1 2 3 Курц А. Л., Брусова Г. П., Демьянович В. М. Раздел III. Свойства одноатомных спиртов. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. ChemNet. Химический факультет МГУ (1999). Архивировано из первоисточника 21 августа 2011. Проверено 28 августа 2009.
- ↑ 1 2 3 Прянишников Н. Д. Практикум по органической химии. — М.: Государственное научно-техническое издательство химической литературы, 1956. — 244 с.
- ↑ 1 2 3 4 5 6 7 8 Smith M. B., March J. March's Advanced Organic Chemistry. — 6th ed. — Hoboken, NJ: Wiley-Interscience, 2007. — P. 576-577. — 2384 p. — ISBN 0471720917
- ↑ Jones R., Pattison J. B. Reaction between unsaturated alcohols and potassium iodide in the presence of polyphosphoric acid // Journal of the Chemical Society C: Organic. — 1969. — № 7. — P. 1046-1047. — DOI:10.1039/J39690001046
- ↑ Deo M. Di, Marcantoni E., Torregiani E. A Simple, Efficient, and General Method for the Conversion of Alcohols into Alkyl Iodides by a CeCl3·7H2O/NaI System in Acetonitrile // Journal of Organic Chemistry. — 2000. — Т. 65. — № 9. — P. 2830–2833. — DOI:10.1021/jo991894c
- ↑ Copenhaver J. E., Whaley A. M. n-Butyl chloride // Organic Syntheses. — 1941. — Т. I. — P. 142.
- ↑ Fuchs R., Cole L. L. The Synthesis of Primary Alkyl Chlorides from Alcohols and Hydrogen Chloride in Hexamethylphosphoric Triamide // Can. J. Chem. — 1975. — Т. 53. — № 23. — P. 3620–3621. — DOI:10.1139/v75-522
- ↑ 1 2 Рахимов А. И. Химия и технология фторорганических соединений // . — М.: Химия, 1986. — С. 134.
- ↑ 1 2 3 Агрономов А. Е. Избранные главы органической химии: Учебное пособие для вузов. — М.: «Химия», 1990. — 560 с. — ISBN 5-7245-0387-5
- ↑ 1 2 Comprehensive Organic Functional Group Transformations / Edited by Alan R. Katritzky, Steven V. Ley, Otto Meth-Cohn, Charles Wayne Rees. — First Edition. — Elsevier, 1995. — Vol. 2. Synthesis: Carbon with One Heteroatom Attached by a Single Bond. — 1441 p. — ISBN 0-08-042323-X
- ↑ 1 2 Кери Ф., Сандберг Р. Углубленный курс органической химии = Advanced organic chemistry. — М.: Химия, 1981. — Т. 2. — 456 с.
- ↑ Аппеля реакция (Appel reaction). Архивировано из первоисточника 21 августа 2011. Проверено 3 июня 2010.
- ↑ Appel Reaction. Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 6 июля 2010.
- ↑ 1 2 3 Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д. Бартона и В. Д. Оллиса. — М.: «Химия», 1982. — Т. 2. — 856 с.
- ↑ Olah G. A., Nojima M., Kerekes I. Synthetic methods and reactions. I. Seleniuum tetrafluoride and its pyridine complex. Convenient fluorinating agents for fluorination of ketones, aldehydes, amides, alcohols, carboxylic acids, and anhydrides (англ.) // Journal of The Americal Chemical Society. — 1974. — Т. 96. — № 3. — С. 925—927.
- ↑ Dinoiu V. Chemical Fluorination of Organic Compounds (англ.) // Revue Roumaine de Chimie. — 2007. — Т. 52. — № 3. — С. 219—234.
- ↑ David A. F., Olofson R. A. A useful conversion of alcohols to alkyl fluorides (англ.) // Tetrahedron Letters. — 2002. — Т. 43. — № 23. — С. 4275—4279.
- ↑ De Luca L., Giacomelli G., Porcheddu A. An Efficient Route to Alkyl Chlorides from Alcohols Using the Complex TCT/DMF (англ.) // Organic Letters. — 2002. — Т. 4. — № 4. — С. 553—555.
- ↑ Snyder D. C. Conversion of Alcohols to Chlorides by TMSCl and DMSO (англ.) // The Journal of Organic Chemistry. — 1995. — Т. 60. — № 8. — С. 2638—2639.
- ↑ Ellwood A. R., Porter M. J. Selective Conversion of Alcohols into Alkyl Iodides Using a Thioiminium Salt (англ.) // The Journal of Organic Chemistry. — 2009. — Т. 74. — № 20. — С. 7982—7985.
- ↑ Нитраты органические // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1992. — Т. 3. — С. 505—506. — ISBN 5-85270-039-8
- ↑ Нитриты органические // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1992. — Т. 3. — С. 518—519. — ISBN 5-85270-039-8
- ↑ Synthesis of Nitrites (англ.). C-O Bond Formation. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 5 июля 2010.
- ↑ 1 2 3 4 Травень В. Ф. Органическая химия: Учебник для вузов: В 2 т. / В. Ф. Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 2. — 582 с. — ISBN 5-94628-172-0
- ↑ Алкилсульфаты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 160—161.
- ↑ Сульфиты органические // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1995. — Т. 4. — С. 921—922. — ISBN 5-85270-092-4
- ↑ Селезнёв Д. В. Синтез и исследование гомолитических превращений алкилгипогалогенитов и алкилнитритов. — Автореферат диссертации. — Уфа: Уфимский ГНТУ, 2002. — С. 5.
- ↑ 1 2 3 Спирты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1992. — Т. 3. — С. 800—804. — ISBN 5-85270-039-8
- ↑ Фосфаты органические // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1999. — Т. 5. — С. 255—256. — ISBN 5-85270-310-9
- ↑ Фосфиты органические // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1999. — Т. 5. — С. 266. — ISBN 5-85270-310-9
- ↑ Бораты органические // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 582.
- ↑ Тиоцианаты органические // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1995. — Т. 4. — С. 1167. — ISBN 5-85270-092-4
- ↑ 1 2 3 Вильямсона синтез // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 710—711.
- ↑ 1 2 Курц А. Л., Брусова Г. П., Демьянович В. М. Простые эфиры. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. ChemNet. Химический факультет МГУ (1999). Архивировано из первоисточника 21 августа 2011. Проверено 4 июня 2010.
- ↑ 1 2 3 Шабаров Ю. С. Органическая химия. Часть 1. Нециклические соединения. — М.: «Химия», 1994. — Т. 1. — С. 180—182. — ISBN 5-7245-0656-4
- ↑ Pines H., Hensel J., Sǐmonik J. Reactions of alcohols: VI. Dehydration of primary alkanols to ethers in a flow system over supported nickel catalysts in the presence of hydrogen. Effect of supports (англ.) // Journal of Catalysis. — 1925. — Т. 24. — № 2. — С. 206—2107.
- ↑ Эфиры простые // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1999. — Т. 5. — С. 1008—1009. — ISBN 5-85270-310-9
- ↑ Fischer Esterification. Fischer-Speier Esterification (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 7 июля 2010.
- ↑ Heteroatom manipulation // Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry / Editor-in-Chief Barry M. Trost, Ian Fleming, Volume editor Ekkehard Winterfeldt. — Pergamon Press, 2005. — Т. 6. — P. 325—327. — 1195 p. — ISBN 0-08-040597-5
- ↑ Steglich Esterification (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 8 июля 2010.
- ↑ 1 2 3 4 Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1987. — Т. 2. — 504 с.
- ↑ Biodiesel Production. Biodiesel Fact Sheets. The National Biodiesel Board. Архивировано из первоисточника 21 августа 2011. Проверено 8 июля 2010.
- ↑ Korus R. A., Hoffman D. S., Barn N., Peterson C. L., Drown D. C. Transesterification Process to Manufacture Ethyl Ester of Rape Oil. Biodiesel Research, Documentation & Articles. Biodiesel Gear! (run by Biodiesel Tecnologies Int'l). Архивировано из первоисточника 21 августа 2011. Проверено 8 июля 2010.
- ↑ Вацуро К. В., Мищенко Г. Л. 5. Айнхорн (Einhorn) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 9.
- ↑ Pinner Reaction (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 6 июля 2010.
- ↑ Teruaki Mukaiyama, Masahiro Usui, Eiichiro Shimada, Kazuhiko Saigo A Convenient Method for the Synthesis of Carboxylic Esters (англ.) // Chemistry Letters. — 1975. — Т. 4. — № 10. — С. 1045—1048.
- ↑ Hua Zhao, Zhiyan Song, Janet V. Cowins, Olarongbe Olubajo. Microwave-Assisted Esterification of N-Acetyl-L-Phenylalanine Using Modified Mukaiyama's Reagents: A New Approach Involving Ionic Liquids (англ.) // International Journal of Molecular Sciences. — 2008. — Т. 9. — № 1. — С. 33—44.
- ↑ Grochowski E., Hilton B. D., Kupper R. J., Michejda C. J. Mechanism of the Triphenylphosphine and Diethyl Azodicarboxylate Induced Dehydration Reactions (Mitsunobu Reaction). The Central Role of Pentavalent Phosphorus Intermediates (англ.) // Journal of The American Chemical Society. — 1982. — Т. 104. — № 24. — С. 6876—6877.
- ↑ Camp D., Jenkins I. D. The Mechanism of the Mitsunobu Esterification Reaction. Part I. The Involvement of Phosphoranes and Oxyphosphonium Salts (англ.) // The Journal of Organic Chemistry. — 1989. — Т. 54. — № 13. — С. 3045—3049.
- ↑ Camp D., Jenkins I. D. The Mechanism of the Mitsunobu Esterification Reaction. Part II. The Involvement of (Acyloxy)alkoxyphosphoranes (англ.) // The Journal of Organic Chemistry. — 1989. — Т. 54. — № 13. — С. 3049—3054.
- ↑ 1 2 3 Cotarca L., Eckert H. Phosgenations – A Handbook. — Weinheim: Wiley-VCH, 2003. — 656 p. — ISBN 3-527-29823-1
- ↑ 1 2 Бюлер К., Пирсон Д. Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — 621 с.
- ↑ Амиламины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 232—233.
- ↑ Тарасевич В. А., Козлов Н. Г. Восстановительное аминирование кислородсодержащих органических соединений // Успехи химии. — 1999. — Т. 68. — № 1. — С. 61—79.
- ↑ N-Алкиланилины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 154.
- ↑ 1 2 3 Матьё Ж., Панико Р., Вейль-Рейналь Ж. Изменение и введение функций в органическом синтезе = L'amenagement fonctionnel en synthese organique / Перевод с французского С. С. Юфита. — М.: «Мир», 1980. — 439 с.
- ↑ Тиолы // Химическая энциклопедия / Главный редактор H. C. Зефиров. — М.: Научное Издательство «Большая Российская Энциклопедия», 1995. — Т. 4. — С. 1138. — ISBN 5-85270-092-4
- ↑ Frank R. L., Smith P. V. The Preparation of Mercaptans from Alcohols (англ.) // Journal of The American Chemical Society. — 1946. — Т. 68. — № 10. — С. 2103—2104.
- ↑ 1 2 3 Бюлер К., Пирсон Д. Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 2. — 592 с.
- ↑ Li J. J., Limberakis C., Pflum D. A. Modern organic synthesis in the laboratory: a collection of standard experimental procedures. — New York: Oxford University Press, Inc, 2007. — P. 31—32. — ISBN 978-0-19-518798-4
- ↑ 1 2 3 Реутов О. А, Курц А. Л., Бутин К. П. Органическая химия. — М.: Издательство МГУ, 1999. — Т. 2. — 624 с. — ISBN 5-211-03491-0
- ↑ Skell P. S., Starer I. Reaction ox Alkoxide with CX2 to Produce Carbonium Ion Intermediates (англ.) // Journal of The Americal Chemical Society. — 1959. — Т. 81. — № 15. — С. 4117—4118.
- ↑ Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д. Бартона и В. Д. Оллиса. — М.: «Химия», 1981. — Т. 1. — 736 с.
- ↑ 1 2 Олефины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1992. — Т. 3. — С. 739. — ISBN 5-85270-039-8
- ↑ Adkins H., Perkins Ph. P. Dehydration of Alcohols over Alumina (англ.) // Journal of The Americal Chemical Society. — 1925. — Т. 47. — № 4. — С. 1163—1167.
- ↑ Lundeen A. J., VanHoozer R. Selective Catalytic Dehydration. Thoria-catalyzed Dehydration of Alcohols (англ.) // The Journal of Organic Chemistry. — 1967. — Т. 32. — № 11. — С. 3386—3389.
- ↑ 1 2 Lazier W. A., Adkins H. Dehydrogenation and Dehydration of Alcohols over a Zinc Oxide Catalyst (англ.) // Journal of The Americal Chemical Society. — 1925. — Т. 47. — № 6. — С. 1719—1722.
- ↑ Komarewsky V. I., Price C. F., Coley J. R. The Hydrogenolysis of Aliphatic Alcohols (англ.) // Journal of The Americal Chemical Society. — 1947. — Т. 69. — № 2. — С. 238—239.
- ↑ Gotoha H., Yamadaa Y., Sato S. Dehydration of 1,3-butanediol Over Rare Earth Oxides (англ.) // Applied Catalysis A: General. — 2010. — Т. 377. — № 1—2. — С. 92—98.
- ↑ Bryant D. E., Kranich W. L. Dehydration of Alcohols Over Zeolite Catalysts (англ.) // Journal of Catalysis. — 1967. — Т. 8. — № 1. — С. 8—13.
- ↑ Pillai C. N., Pines H. Alumina: Catalyst and Support. XI. Mechanism of Dehydration of Aliphatic Alcohols and the Formation of Cyclopropanes during Dehydration (англ.) // Journal of The Americal Chemical Society. — 1961. — Т. 83. — № 15. — С. 3274—3279.
- ↑ Burgess E. M., Penton Jr. H. R. , Taylor E. A. Thermal Reactions of Alkyl N-carbomethoxysulfamate Esters (англ.) // The Journal of Organic Chemistry. — 1973. — Т. 38. — № 1. — С. 26—31.
- ↑ 1 2 Mundy B. P., Ellerd M. G., Favaloro, Jr. F. G. Name Reactions and Reagents in Organic Synthesis. — Second edition. — Hoboken: John Wiley & Sons, Inc, 2005. — 882 p. — ISBN 0-471-22854-0
- ↑ Martin J. C., Arhart R. J. Sulfuranes. II. Isolation and Characterization of a Crystalline Dialkoxydiarylsulfurane (англ.) // Journal of The American Chemical Society. — 1971. — Т. 93. — № 9. — С. 2341.
- ↑ Реакция Чугаева. Именные органические реакции. Иркутский государственный университет. Химический факультет. Архивировано из первоисточника 21 августа 2011. Проверено 22 мая 2010.
- ↑ 1 2 Hudlický M. Oxidation in Organic Chemistry. — ACS monograph 186. — Washington: American Chemical Society, 1990. — 434 p. — ISBN 0-8412-1780-7
- ↑ Робертс Дж., Касерио М. Основы органической химии = Basic principles of organic chemistry / Под редакцией академика Несмеянова А. Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — 843 с.
- ↑ Jones Oxidation (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 1 июля 2010.
- ↑ Fuhrhop J.-H., Li G. Organic Synthesis: Concepts and Methods. — Third Edition. — Wiley-VCH, 2003. — P. 190. — ISBN 978-35273-0272-7
- ↑ Sarett Reagent (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 1 июля 2010.
- ↑ Collins Reagent (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 1 июля 2010.
- ↑ Jain S., Hiran B. L., Bhatt C. V. Oxidation of Some Aliphatic Alcohols by Pyridinium Chlorochromate - Kinetics and Mechanism (англ.) // E-Journal of Chemistry : web publication. — 2009. — Т. 6. — № 1. — С. 237—246.
- ↑ Banerji K. Kinetics and Mechanism of the Oxidation of Alcohols by Pyridinium Chlorochromate (англ.) // Bulletin of The Chemical Society of Japan. — 1978. — Т. 51. — № 9. — С. 2732—2734.
- ↑ Oxidatgion // Comprehensive Organic Synthesis: Selectivity, Strategy, and Efficiency in Modern Organic Chemistry / Editor-in-chief Barry M. Trost, Deputy editor-in-chief Ian Fleming, Volume editor Steven V. Ley. — Fifth impression. — Oxford: Pergamon Press, 2005. — Т. 7. — P. 267—278. — ISBN 0-08-040598-3
- ↑ 1 2 Вацуро К. В., Мищенко Г. Л. 97. Болл — Гудвин — Мортон (Ball — Goodwin — Morton) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 68—69.
- ↑ Banerji K. K. Kinetics and Mechanism of the Oxidation of Aliphatic Alcohols by Acid Permanganate (англ.) // Bulletin of The Chemical Society of Japan. — 1973. — Т. 46. — № 11. — С. 3623—3624.
- ↑ Josea N., Senguptaa S., Basuand J. K. Selective Production of Benzaldehyde by Permanganate Oxidation of Benzyl Alcohol Using 18-crown-6 as Phase Transfer Catalyst (англ.) // Journal of Molecular Catalysis A: Chemical. — 2009. — Т. 309. — № 1—2. — С. 153—158.
- ↑ Кетоны // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1990. — Т. 2. — С. 746—747. — ISBN 5-85270-035-5
- ↑ Вацуро К. В., Мищенко Г. Л. 410. Муро — Миньонак (Moureau — Mignonac) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 286.
- ↑ Вацуро К. В., Мищенко Г. Л. 3. Адкинс — Питерсон (Adkins — Peterson) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 8.
- ↑ Беспалов П. Каталитическое окисление этанола. Органическая химия. Видеоопыты. Единая коллекция цифровых образовательных ресурсов. Архивировано из первоисточника 21 августа 2011. Проверено 6 июля 2010.
- ↑ Беспалов П. Окисление этилового спирта оксидом меди (II). Органическая химия. Видеоопыты. Единая коллекция цифровых образовательных ресурсов. Архивировано из первоисточника 21 августа 2011. Проверено 6 июля 2010.
- ↑ Lippits M. J., Nieuwenhuysa B. E. Direct conversion of ethanol into ethylene oxide on copper and silver nanoparticles: Effect of addition of CeOx and Li2O // Catalysis Today. — Available online, 2010. — № 24 April 2010.
- ↑ Velusamy S., Punniyamurthy T. Novel Vanadium-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones under Atmospheric Oxygen // Organic Letters. — 2004. — Т. 6. — № 2. — P. 217—219.
- ↑ Qiana G., Zhaoa Rui., Lua G., Qia Y., Suo J. Partial Oxidation of Alcohols to Aldehydes and Ketones Under Mild Conditions // Synthetic Communications. — 2004. — Т. 34. — № 10. — P. 1753—1758.
- ↑ Nishimura T., Onoue T., Ohe K., Uemura S. Pd(OAc)2-catalyzed oxidation of alcohols to aldehydes and ketones by molecular oxygen // Tetrahedron Letters. — 1998. — Т. 39. — № 33. — P. 6011—6014.
- ↑ Micovic V. M., Mamuzi R. I., Jeremi D., Mihailovic M. Lj. Reactions with lead tetraacetate—I // Tetrahedron. — 1964. — Т. 20. — № 10. — P. 2279—2287. — DOI:10.1016/S0040-4020(01)97615-X
- ↑ Вацуро К. В., Мищенко Г. Л. 341. Криге (Criegee) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 240.
- ↑ Вацуро К. В., Мищенко Г. Л. 387. Маурер — Дрефаль (Maurer — Drefahl) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 270.
- ↑ Nwaukwa S. O., Keehn P. M. The oxidation of alcohols and ethers using calcium hypochlorite [Ca(OCl)2] // Tetrahedron. — 1982. — Т. 23. — № 1. — P. 35—38. — DOI:10.1016/S0040-4039(00)97525-7
- ↑ Kooti M., Tarassoli A., Javadi H., Jofri M. A Facile Conversion of Alcohols to Esters Mediated by Potassium Ferrate // E-Journal of Chemistry. — 2008. — Т. 5. — № 4. — P. 718—722.
- ↑ Kakis F. J. , Fetizon M., Douchkine N., Golfier M., Mourgues Ph., Prange T. Mechanistic Studies Regarding the Oxidation of Alcohols by Silver Carbonate on Celite // The Journal of Organic Chemistry. — 1974. — Т. 39. — № 4. — P. 523—533.
- ↑ Pfitzner K. E., Moffatt J. G. A New and Selective Oxidation of Alcohols (англ.) // Journal of The Americal Chemical Society. — 1963. — Т. 85. — № 19. — С. 3027—3028.
- ↑ 1 2 Pfitzner K. E., Moffatt J. G. Sulfoxide-Carbodiimide Reactions. II. Scope of the Oxidation Reaction (англ.) // Journal of The Americal Chemical Society. — 1965. — Т. 87. — № 24. — С. 5670—5678.
- ↑ 1 2 3 Albright J. D., Goldman L. Dimethyl Sulfoxide-Acid Anhydride Mixtures. New Reagents for Oxidation of Alcohols (англ.) // Journal of The Americal Chemical Society. — 1965. — Т. 87. — № 18. — С. 4214—4216.
- ↑ Pfitzner K. E., Moffatt J. G. Sulfoxide-Carbodiimide Reactions. I. A Facile Oxidation of Alcohols (англ.) // Journal of The Americal Chemical Society. — 1965. — Т. 87. — № 24. — С. 5661—5670.
- ↑ 1 2 3 4 Tojo G., Fernández M. Oxidation of Alcohols to Aldehydes and Ketones. — First Edition. — New York: Springer, 2006. — 375 p. — ISBN 0-387-23607-4
- ↑ Onodera K., Hirano S., Kashimura N. Oxidation of Carbohydrates with Dimethyl Sulfoxide Containing Phosphorus Pentoxide (англ.) // Journal of The Americal Chemical Society. — 1965. — Т. 87. — № 20. — С. 4651—4652.
- ↑ Palomo C., Aizpurua J. M., Urchegui R, Garcia J.M. A facile access to peptides containing D-α-methyl β-alkylserines by coupling of α-branched leuchs anhydrides with α-amino esters (англ.) // Journal of the Chemical Society, Chemical Communications. — 1995. — № 22. — С. 2327—2328. — DOI:10.1039/C39950002327
- ↑ Parikh J. R., Doering W. v. E. Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide (англ.) // Journal of The Americal Chemical Society. — 1967. — Т. 89. — № 21. — С. 5505—5507.
- ↑ 1 2 Mancuso A. J., Huang Shui-Lung, Swern D. Oxidation of long-chain and related alcohols to carbonyls by dimethyl sulfoxide "activated" by oxalyl chloride (англ.) // The Journal of Organic Chemistry. — 1978. — Т. 43. — № 12. — С. 2480—2482.
- ↑ Swern Oxidation (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 1 июля 2010.
- ↑ Mancuso A. J., Brownfain D. S., Swern D. Structure of the dimethyl sulfoxide-oxalyl chloride reaction product. Oxidation of heteroaromatic and diverse alcohols to carbonyl compounds (англ.) // The Journal of Organic Chemistry. — 1979. — Т. 44. — № 23. — С. 4148—4150.
- ↑ Corey-Kim Oxidation (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 4 июля 2010.
- ↑ 1 2 Corey E. J., Kim C. U. New and highly effective method for the oxidation of primary and secondary alcohols to carbonyl compounds (англ.) // Journal of The Americal Chemical Society. — 1972. — Т. 94. — № 21. — С. 7586—7587.
- ↑ Meerwein H., Schmidt R. Ein neues Verfahren zur Reduktion von Aldehyden und Ketonen (нем.) // Justus Liebigs Annalen der Chemie. — 1925. — Т. 444. — № 1. — С. 221—238.
- ↑ Ponndorf W. Der reversible Austausch der Oxydationsstufen zwischen Aldehyden oder Ketonen einerseits und primären oder sekundären Alkoholen anderseits (нем.) // Angewandte Chemie. — 1925. — Т. 39. — № 5. — С. 138—143.
- ↑ Meerwein-Ponndorf-Verley Reduction (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 4 июля 2010.
- ↑ Oppenauer Oxidation (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 4 июля 2010.
- ↑ Ли Дж. Оппенауэр (Oppenauer). Окисление // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В. М. Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 253. — ISBN 5-94774-368-X
- ↑ Narasaka K., Morikawa A., Saigo K., Mukaiyama T. Efficient Methods for Oxidation of Alcohols (англ.) // Bulletin of the Chemical Society of Japan. — 1977. — Т. 50. — № 10. — С. 2773—2776.
- ↑ Dess D. B., Martin J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones (англ.) // The Journal of Organic Chemistry. — 1983. — Т. 48. — № 22. — С. 4155—4156.
- ↑ Ireland R. E. , Liu L. An improved procedure for the preparation of the Dess-Martin periodinane (англ.) // The Journal of Organic Chemistry. — 1993. — Т. 58. — № 10. — С. 2899.
- ↑ Meyer S. D., Schreiber S. L. Acceleration of the Dess-Martin Oxidation by Water (англ.) // The Journal of Organic Chemistry. — 1994. — Т. 59. — № 24. — С. 7549—7552.
- ↑ 1 2 Hypervalent Iodine Compounds (англ.). Oxidizing Agents. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 4 июля 2010.
- ↑ Anelli P. L., Biffi C., Montanari F., Quici S. Fast and Selective Oxidation of Primary Alcohols to Aldehydes or to Carboxylic Acids and of Secondary Alcohols to Ketones Mediated by Oxoammonium Salts under Two-Phase Conditions (англ.) // The Journal of Organic Chemistry. — 1987. — Т. 52. — № 12. — С. 2559.
- ↑ Angelin M., Hermansson M., Dong H., Ramström O. Direct, Mild, and Selective Synthesis of Unprotected Dialdo-Glycosides. Abstracts. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 11 августа 2010.
- ↑ TEMPO, 2,2,6,6-Tetramethylpiperidinyloxy. Oxidizing Agents. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 11 августа 2010.
- ↑ Альдегиды // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 198.
- ↑ 1 2 3 4 5 Шелдон Р. А. Химические продукты на основе синтез-газа = Chemicals from Synthesis Gas / Пер. с англ./Под ред. С.М.Локтева. — М.: «Химия», 1987. — 248 с.
- ↑ Matsumura Y., Hashimoto K., Yoshida S. Selective Dehydrogenation of Ethanol over Highly Dehydrated Silica (англ.) // Journal of Catalysis. — 1989. — Т. 117. — № 1. — С. 4026—4033.
- ↑ Dunbar R. E., Arnold M. R. Catalytic Dehydrogenation of Primary and Secondary Alcohols with Copper-Chromium Oxide (англ.) // The Journal of Organic Chemistry. — 1945. — Т. 10. — № 6. — С. 501—504.
- ↑ Zhang J., Gandelman M., Shimon L. J. W, Rozenberg H., Milstein D. Electron-Rich, Bulky Ruthenium PNP-Type Complexes. Acceptorless Catalytic Alcohol Dehydrogenation (англ.) // Organometallics. — 2004. — Т. 23. — № 17. — С. 4026—4033.
- ↑ Zhang J., Leitus G., Ben-David Y., Milstein D. Facile Conversion of Alcohols into Esters and Dihydrogen Catalyzed by New Ruthenium Complexes (англ.) // Journal of American Chemical Society. — 2005. — Т. 127. — № 31. — С. 10840—10841.
- ↑ Gunanathan C., Shimon L. J. W., Milstein D. Direct Conversion of Alcohols to Acetals and H2 Catalyzed by an Acridine-Based Ruthenium Pincer Complex (англ.) // Journal of American Chemical Society. — 2009. — Т. 131. — № 9. — С. 3146—3147.
- ↑ Hutkins R. W. Microbiology and Technology of Fermented Foods. — First edition. — Ames: Blackwell Publishing, 2006. — P. 401—403. — 473 p. — ISBN 0-8138-0018-8
- ↑ Gluconobacter oxydans has the ability to oxidize ethanol to acetic acid (англ.). Bacteria Genomes. European Bioinformatics Institute. Архивировано из первоисточника 21 августа 2011. Проверено 10 апреля 2011.
- ↑ Covert L. W., Connor R., Adkins H. The Use of Nickel as a Catalyst for Hydrogenation. II (англ.) // Journal of The Americal Chemical Society. — 1932. — Т. 54. — № 4. — С. 1651—1663.
- ↑ Connor R., Adkins H. Hydrogenolysis of Oxygenated Organic Compounds (англ.) // Journal of The Americal Chemical Society. — 1932. — Т. 54. — № 12. — С. 4678—4690.
- ↑ 1 2 Notheisz F., Bartók M. Hydrogenolysis of C−O, C−N and C—X Bonds // Fine Сhemicals through Heterogenous Catalysis / Edited by Roger A. Sheldon, Herman van Bekkum. — Wiley-VCH, 2001. — P. 415. — ISBN 3-527-29951-3
- ↑ 1 2 3 Reduction // Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry / Editor-in-Chief Barry M. Trost, Volume editor Ian Fleming. — Pergamon Press, 2005. — Т. 8. — P. 814—815. — 1140 p. — ISBN 0-08-040599-1
- ↑ Zhang Shu-Yu, Tu Yong-Qiang, Fan Chun-An, Jiang Yi-Jun, Shi Lei, Cao Ke, Zhang En. Cross-Coupling Reaction between Alcohols through sp3 CH Activation Catalyzed by a Ruthenium/Lewis Acid System (англ.) // Chemistry - A European Journal. — 2008. — Т. 14. — № 33. — С. 10201—10205.
- ↑ Barton-McCombie Reaction. Barton Deoxygenation (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 3 июня 2010.
- ↑ 1 2 Дядченко В. П., Трушков И. В., Брусова Г. П. Синтетические методы органической химии. Части 1-2. — М.: МГУ им. М. В. Ломоносова, 2004. — С. 70—73.
- ↑ Reppe W., Kröper H., Kutepow N., Pistor H. J. Carbonylierung III. Umsetzung von Alkoholen und offenen Äthern mit Kohlenoxyd zu Carbonsäuren (нем.) // Justus Liebigs Annalen der Chemie. — Т. 582. — № 1. — С. 72—86.
- ↑ Караханов Э. А. Синтез-газ как альтернатива нефти. II. Метанол и синтезы на его основе // Соросовский образовательный журнал. — 1997. — № 12. — С. 68.
- ↑ 1 2 Брук Л. Г., Ошанина И. В., Городский С. Н., Темкин О. Н. Окислительное карбонилирование и сопряженные процессы с участием оксида углерода, катализируемые комплексами палладия // Российский химический журнал. — 2006. — Т. L. — № 4. — С. 104.
- ↑ Пинаколиновая и ретропинаколиновая перегруппировки // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1992. — Т. 3. — С. 1023—1024. — ISBN 5-85270-039-8
- ↑ Вагнера — Меервейна перегруппировки // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 659—660.
- ↑ Tsuji J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis. — New York: John Wiley & Sons Ltd, 1995. — P. 31. — ISBN 0-471-95483-7
- ↑ 1 2 3 4 Ушакова И. П., Брагина Н. А., Миронов А. Ф. Защитные группы в тонком органическом синтезе. Учебное пособие. — М.: МИТХТ им. М. В. Ломоносова, 2004. — С. 13-18.
- ↑ Greene Th. W., Wuts P. G. M. Protective Groups in Organic Synthesis. — Third Edition. — New York: John Wiley & Sons, Inc, 1999. — P. 23—244. — 748 p. — ISBN 0-471-16019-9
- ↑ McMurry J. Organic chemistry. — Seven edition. — Thomson, 2008. — ISBN 0-495-11258-5
- ↑ 1 2 3 4 5 6 Защитные группы в органической химии / Под редакцией Дж. МакОми, перевод с английского. — М.: «Мир», 1976. — 392 с.
Литература
- Англоязычная
- Alcohols / Volume Editor: Prof. Jonathan Clayden. — Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. — Georg Thieme Verlag, 2008. — Т. 36. — 1294 p. — (Category 5: Compounds with One Carbon-Heteroatom Bonds). — ISBN 978-1-588-90527-7
- Mellan I. Polyhydric Alcohols. — Spartan Books. — 1962. — 208 p.
- Monick J. A. Alcohols: Their Chemistry, Properties, and Manufacture. — Reinhold, 1968. — 594 p. — ISBN 0-442-15601-4
- Monohydric Alcohols: Manufacture, Applications, and Chemistry: based on a symposium / Editor: Edward J. Wickson. — American Chemical Society, 1981. — 222 p. — (ACS symposium series (volume 159)). — ISBN 0-841-20637-6
- Otera J., Nishikido J. Esterification: Methods, Reactions, and Applications. — Second edition. — Weinheim: Wiley-VCH, 2010. — 374 p. — ISBN 978-3-527-32289-3
- Tojo G., Fernández M. Oxidation of Alcohols to Aldehydes and Ketones. — First Edition. — New York: Springer, 2006. — 375 p. — ISBN 0-387-23607-4
- Weissermel K., Arpe H-J. Alcohols // Industrial organic chemistry. — 4th ed. — Weinheim: Wiley-VCH, 2003. — P. 193—266. — ISBN 978-3-527-30578-0
- Русскоязычная
- Высшие жирные спирты (области применения, методы производства, физико-химические свойства) / Под редакцией С. М. Локтева. — М.: «Химия», 1970. — 329 с.
- Курц А. Л., Брусова Г. П., Демьянович В. М. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. Учебные материалы. Органическая химия. ChemNet. Химический факультет МГУ (1999). Архивировано из первоисточника 21 августа 2011. Проверено 10 июля 2010.
Категории:- Спирты
- Химические реакции
Wikimedia Foundation. 2010.
Полезное
Смотреть что такое "Химические свойства спиртов" в других словарях:
Химические свойства — свойства веществ (химических элементов, простых веществ и химических соединений), имеющие отношение к химическим процессам, то есть проявляемые в процессе химической реакции. К химическим свойствам относятся способность реагировать с другими… … Википедия
Свойства фосфолипидов — Фосфолипид Фосфолипиды сложные липиды, сложные эфиры многоатомных спиртов и высших жирных кислот. Содержат остаток фосфорной кислоты и соединенную с ней добавочную группу атомов различной химической природы. Содержание 1 Классификация… … Википедия
Химические формулы — Химическая формула отражение информации о составе и структуре веществ с помощью химических знаков, чисел и разделяющих знаков скобок. В настоящее время различают следующие виды химических формул: Простейшая формула. Может быть получена опытным… … Википедия
Ртуть, её свойства, соединения и получение — (живое серебро, Hydrargirum, Quecksilber, mercure), Hg принадлежит к числу 7 металлов, известных в глубокой древности: золото, серебро, медь, железо, свинец, олово и Р. По сравнению с остальными 6 металлами человек, по всей вероятности,… … Энциклопедический словарь Ф.А. Брокгауза и И.А. Ефрона
Формулы химические — Химическая формула отражение информации о составе и структуре веществ с помощью химических знаков, чисел и разделяющих знаков скобок. В настоящее время различают следующие виды химических формул: Простейшая формула. Может быть получена опытным… … Википедия
Спирты — Отличительная особенность спиртов гидроксильная группа при насыщенном атоме углерода на рисунке выделена красным (кислород) и серым цветом (водород). Спирты (от лат. … Википедия
Одноатомные спирты — Спирты (устар. алкоголи) органические соединения, содержащие одну или несколько гидроксильных групп (гидроксил, OH), непосредственно связанных с атомом углерода в углеводородном радикале. Общая формула простых предельных (ациклических) спиртов… … Википедия
Одноатомный спирт — Спирты (устар. алкоголи) органические соединения, содержащие одну или несколько гидроксильных групп (гидроксил, OH), непосредственно связанных с атомом углерода в углеводородном радикале. Общая формула простых предельных (ациклических) спиртов… … Википедия
Алкены — У этого термина существуют и другие значения, см. Алкен (значения). Пространственная структура этилена. Алкены (олефины, этиленовые углеводороды) ациклические непредельные углеводороды … Википедия
Окись этилена — Окись этилена … Википедия
 18+© Академик, 2000-2025
18+© Академик, 2000-2025- Обратная связь: Техподдержка, Реклама на сайте
Экспорт словарей на сайты, сделанные на PHP, Joomla, Drupal, WordPress, MODx.

![\mathsf{6(CH_3)_3COH+2Al}\rightarrow\mathsf{2[(CH_3)_3CO]_3Al+3H_2}](6f8bbd36031d713fb15ad6826bb59b14.png)

![\mathsf{CH_3OH+AlCl_3}\rightarrow\mathsf{[CH_3OH]^+AlCl_3^-}](8d4c9d85345978f4706f80325d8980b6.png)
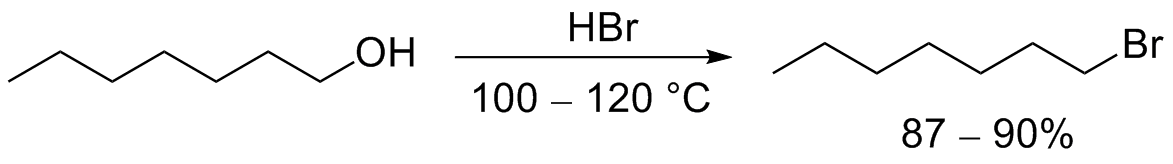














![\mathsf{CCl_3COC(O)COCl_3}\ \xrightarrow{\mathrm{KF/CH_3CN}}\ \ \mathsf{COF_2}\ \xrightarrow[\mathrm{-HF}]{\mathrm{+ROH;\ KF}}\ \ \mathsf{ROC(O)F\ \xrightarrow[\mathrm{-CO_2}]{\mathrm{HBGF}}\ \ \mathsf{RF}}](e9492816c13d3253f906380ae9fc3913.png)


![\mathsf{RR'OH}\ \xrightarrow[\mathrm{THF}]{\mathrm{[CH_3-S-CH=N^+(CH_3)_2]I^-}}\ \ \mathsf{RR'I}\](3c3569c23f6aeb5a3feaa2bbf4bd0e6d.png)




![\mathsf{ROH+SOCl_2+C_5H_5N}\ \xrightarrow{}\ \ \mathsf{RO\!\!-\!\!SOCl+[C_5H_5NH]Cl}\ \xrightarrow{\mathrm{+ROH}}\ \ \mathsf{ROS(O)OR}\](3d7b9b06aa7cf2324e244ea1941a5b3e.png)













![\mathsf{2ROH}\ \xrightarrow[\mathrm{160-190\ ^\circ C}]{\mathrm{Ni-Al_2O_3-SiO_2,\ H_2}}\ \ \mathsf{ROR+H_2O}\](a1c7b453276aa48acd5eaad1fb3b56ee.png)

![\mathsf{RCH\!\!=\!\!CH_2+CH_3OH}\ \xrightarrow[\mathrm{H_3PO_4}]{\mathrm{>100\ ^\circ C}}\ \ \mathsf{RCH_2\!\!-\!\!C(CH_3)OCH_3}\](64df9f74d2cae8c640e465cb2860a6fa.png)







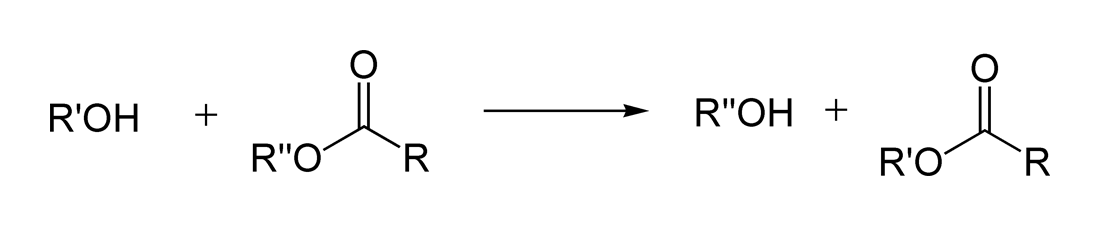


![\mathsf{R^'COCl+C_5H_5N} \rightarrow \mathsf{[C_6H_5N^+\!\!\!-\!\!COR^'Cl^-]}\ \xrightarrow{\mathrm{ROH}} \ \mathsf{R^'COOR}](0e105e33bde91a23f9e6e0265d422baa.png)

![\mathsf{RC}\!\!\equiv\!\!\mathsf{N+HCl} \rightarrow \mathsf{[RC}\!\!\equiv\!\!\mathsf{N^+HCl^-]}\ \xrightarrow{\mathrm{ROH}} \ \mathsf{R(OR)C\!\!=\!\!NH_2^+Cl^-}](e1cb5340b4cf5696762f3ef6f9182ab7.png)







![\mathsf{2C_2H_5OH+H_2SO_4}\ \xrightarrow{}\ \ \mathsf{C_2H_5O\!\!-\!\!SO_2\!\!-\!\!OC_2H_5+2H_2O}\ \xrightarrow[\mathrm{-H_2SO_4}]{\mathrm{2NH_3}}\ \ \mathsf{2C_2H_5NH_2}\ \xrightarrow[\mathrm{-H_2SO_4}]{\mathrm{(C_2H_5O)_2SO_2}}\ \ \mathsf{2(C_2H_5NH)_2}](daab80a8386165474b3deac7523e6656.png)
![\mathsf{ROH+NH_3}\ \xrightarrow[\mathrm{-H_2O}]{\mathrm{Al_2O_3}}\ \ \mathsf{RNH_2+R_2NH+R_3N}](61a06535dd849236eccba5eb9565fbaf.png)
![\mathsf{RCH_2OH}\ \xrightarrow[\mathrm{-H_2}]\ \ \mathsf{RCHO}\ \xrightarrow[\mathrm{-H_2O}]{\mathrm{NH_3}}\ \ \mathsf{RCH\!\!=\!\!NH}\ \xrightarrow[\mathrm{Ni}]{\mathrm{H_2}}\ \ \mathsf{RCH_2NH_2}\ \xrightarrow{\mathrm{RCH\!=\!NH}}\ \ \mathsf{(RCH_2)_2NH}\ \xrightarrow{\mathrm{RCH\!=\!NH}}\ \ \mathsf{(RCH_2)_3N}](618bf70b6de35757cf427e53a44cc7ad.png)
![\mathsf{3CH_3OH+2C_6H_5NH_2}\ \xrightarrow[\mathrm{-H_2O}]{\mathrm{H_3PO_4}}\ \ \mathsf{C_6H_5NHCH_3+C_6H_5N(CH_3)_2}](447fa6c456d4347278df43200b4191ac.png)



![\mathsf{2CH_3OH+H_2SO_4}\ \xrightarrow{}\ \ \mathsf{CH_3O\!\!-\!\!SO_2\!\!-\!\!OCH_3+2H_2O}\ \xrightarrow[\mathrm{-Na_2SO_4}]{\mathrm{2NaCN}}\ \ \mathsf{2CH_3CN}](0915975afe6ac062c83149660d1c3e5c.png)






![\mathsf{RCH_2CH_2OH}\ \xrightarrow[\mathrm{330-420\ ^\circ C}]{\mathrm{ThO_2}}\ \ \mathsf{RCH\!\!=\!\!CH_2+H_2O}](106e78a1b01950e7a84cea4353dffcf8.png)

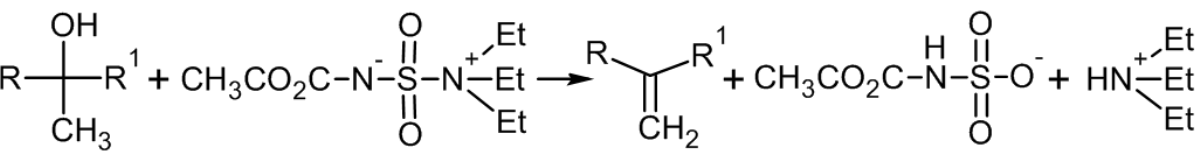








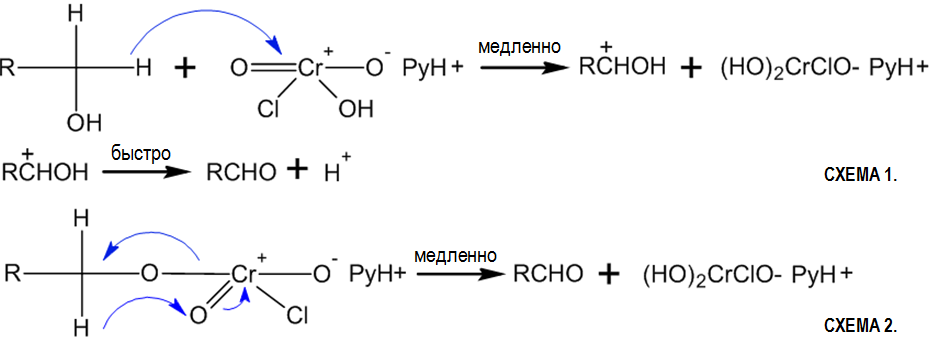
![\mathsf{CH_2\!\!=\!\!CH\!\!-\!\!CH_2OH}\ \xrightarrow[\mathrm{MnO_2}]{\mathrm{20\ ^\circ C}}\ \ \mathsf{CH_2\!\!=\!\!CH\!\!-\!\!CHO}\](e225b7ddb6966db9aa1fe99018b717e2.png)
![\mathsf{CH_2\!\!=\!\!CH\!\!-\!\!CHOH\!\!-\!\!CH_3}\ \xrightarrow[\mathrm{MnO_2}]{\mathrm{20\ ^\circ C}}\ \ \mathsf{CH_2\!\!=\!\!CH\!\!-\!\!C(O)\!\!-\!\!CH_3}\](8a229452273e769585b271ad6a28bfe4.png)

![\mathsf{R\!\!-\!\!CHOH\!\!-\!\!R'}\ \xrightarrow[\mathrm{Ag}]{\mathrm{O_2,\ 360-400\ ^\circ C}}\ \ \mathsf{R\!\!-\!\!CO\!\!-\!\!R'}\](ffa4a8c76742d7de0610e6e5c8ca241f.png)
![\mathsf{2CH_3OH+O_2}\ \xrightarrow[\mathrm{Fe_2O_3}]{\mathrm{250-400\ ^\circ C}}\ \ \mathsf{2CH_2O+2H_2O}\](724f00fdb38c10db57d04b9db534a5be.png)
![\mathsf{2C_2H_5OH+O_2}\ \xrightarrow[\mathrm{Cr_2O_3}]{\mathrm{T^\circ}}\ \ \mathsf{2CH_3CHO+2H_2O}\](4426c3defdc9b3b8f826638b6f3fb8b0.png)
![\mathsf{2C_2H_5OH+O_2}\ \xrightarrow[\mathrm{Ag/Li_2O/Al_2O_3}]{\mathrm{200\ ^\circ C}}\ \ \mathsf{2(CH_2CH_2)O+2H_2O}\](678000afd8f7865380c87e1e5cfa9e49.png)






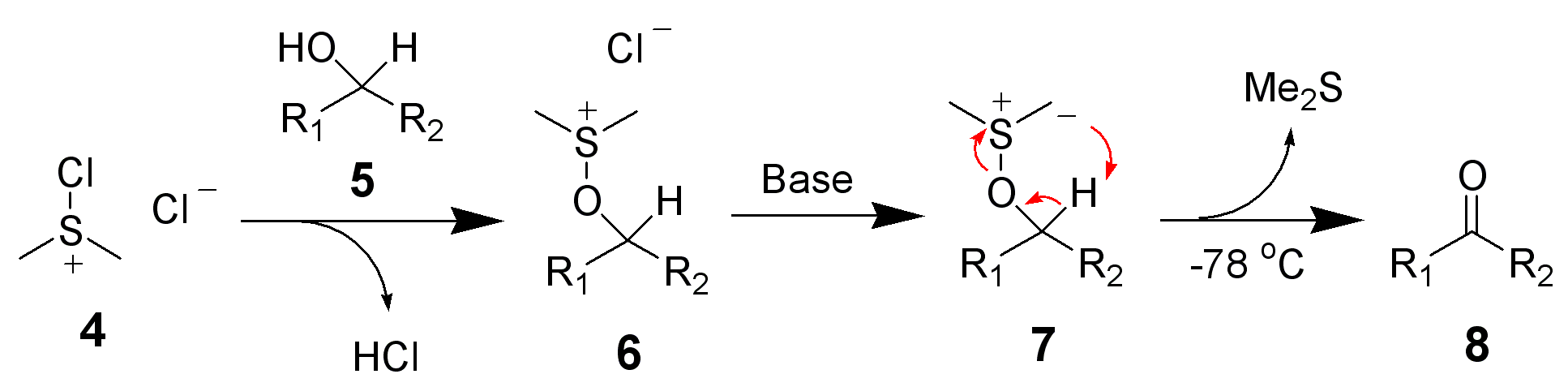

![\mathsf{(CH_3)_2S+Cl_2}\ \xrightarrow[\mathrm{0\ ^\circ C}]{\mathrm{CCl_4}}\ \ \mathsf{(CH_3)_2S^+ClCl^-}](256a4154b0d5ab344aa2f16decf9428d.png)
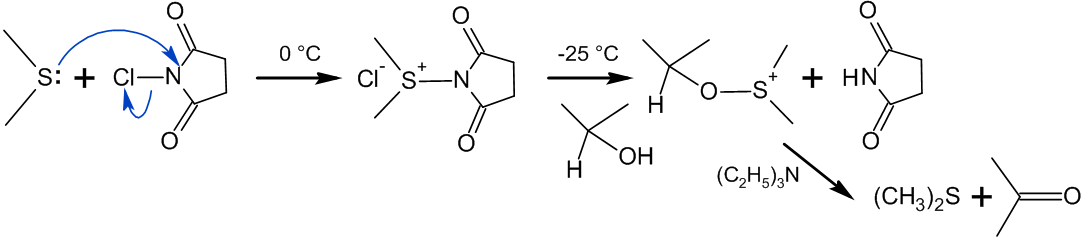


![\mathsf{RR'CH\!\!-\!\!OH}\ \xrightarrow[\mathrm{-C_3H_7OH}]{\mathrm{C_3H_7MgBr}}\ \ \mathsf{[RR'CH\!\!-\!\!OMgBr]}\ \xrightarrow{\mathrm{C_5H_{10}N-C(O)-N=N-C(O)NC_5H_{10}}}\ \ \mathsf{RR'C\!\!=\!\!O}](ab37e35227587b9f20213a02fb2e084b.png)


![\mathsf{RCH_2OH}\ \xrightarrow[\mathrm{NaHCO_3,\ KBr,\ CH_2Cl_2-H_2O}]{\mathrm{4-CH_3O-TEMPO,\ NaOCl}}\ \mathsf{RCHO+H_2O}](9b00c005b7714deac5e70993f5187149.png)





![\mathsf{RR'CHOH}\ \xrightarrow{\mathrm{[HRuClN_2]-complex}}\ \ \mathsf{RR'C\!\!=\!\!O+H_2}](8bec344d2710a53faad6b061b7313e3e.png)
![\mathsf{2RCH_2OH}\ \xrightarrow{\mathrm{[HRu(CO)]-complex}}\ \ \mathsf{RCOOCH_2R+2H_2}](16794b19b8828f23977fbe0115ac462e.png)
![\mathsf{2RCH_2CH_2OH}\ \xrightarrow[\mathrm{base}]{\mathrm{[RuHCl]-complex}}\ \ \mathsf{RCH_2CH_2OC(O)CH_2R+2H_2}](63a67afbf04a5b06f5a56ffd60e8dc05.png)
![\mathsf{2RCH_2CH_2OH}\ \xrightarrow[\mathrm{-H_2O}]{\mathrm{[RuHCl]-complex}}\ \ \mathsf{RCH_2CH(OCH_2CH_2R)_2+H_2}](d1f2da08b60c7ba0839bd20226cca162.png)
![\mathsf{2CH_3CH_2OH+PQQ}\ \xrightarrow[\mathrm{}]{\mathrm{ADH}}\ \ \mathsf{2CH_3CHO+PQQH_2}](5bb28fa632e368e6a469e530faaaba5e.png)
![\mathsf{CH_3CHO+PQQ+H_2O}\ \xrightarrow[\mathrm{}]{\mathrm{ALDH}}\ \ \mathsf{CH_3COOH+PQQH_2}](131cdfd23b81ca89f9f57f20ef2b5368.png)



![\mathsf{CH_2OH\!\!-\!\!CHOH\!\!-\!\!CH_2OH+H_2}\ \xrightarrow[\mathrm{200\ ^\circ C, \ 17\ MPa}]{\mathrm{Ni}}\ \ \mathsf{CH_3\!\!-\!\!CHOH\!\!-\!\!CH_2OH+H_2O}](57e0560bc46e0a8a6bc7f19ae5f0213d.png)





















![\mathsf{2C_2H_5OH+CO+2CuCl_2}\ \xrightarrow[\mathrm{25\ ^\circ C}]{\mathrm{PdCl_2}}\ \ \mathsf{(C_2H_5O)_2C\!\!=\!\!O+2CuCl+2HCl}](5b3595a45f443a2742869aa2235282cf.png)











![\mathsf{R\!\!-\!\!OH+(CH_3)_3SiCl+(CH_3CH_2)_3N}\ \xrightarrow\ \ \mathsf{R\!\!-\!\!O\!\!-\!\!Si(CH_3)_3+[(CH_3CH_2)_3NH]Cl}](34aae052326911c028d2c8236408ddf8.png)




![\mathsf{R\!\!-\!\!OH+(CH_3)_2CH\!\!=\!\!CH_2}\ \xrightarrow[]{\mathrm{H_2SO_4}}\ \mathsf{R\!\!-\!\!OCH(CH_3)_3}](cadaefc39782b859e15661078e99e4f3.png)
![\mathsf{R\!\!-\!\!OH+ BrCH_2\!\!-\!\!CH\!\!=\!\!CH_2}\ \xrightarrow[\mathrm{-NaBr;\ -H_2O}]{\mathrm{NaOH}}\ \mathsf{R\!\!-\!\!OCH\!\!=\!\!CH_2}](60f2bd4f2a011fd5612306c6756cae36.png)
![\mathsf{R\!\!-\!\!OH+ ClCH_2\!\!-\!\!C_6H_5}\ \xrightarrow[\mathrm{-KCl;\ -H_2O}]{\mathrm{KOH}}\ \mathsf{R\!\!-\!\!OCH_2C_6H_5}](4fb8f00cdfe682302a49463b83991b69.png)
![\mathsf{R\!\!-\!\!OH+ ClC(C_6H_5)_3}\ \xrightarrow[\mathrm{-HCl}]{\mathrm{C_5H_5N}}\ \mathsf{R\!\!-\!\!OC(C_6H_5)_3}](7f9d9859d10b76ef2a553d8efb3587b6.png)
![\mathsf{R\!\!-\!\!OH+(CH_3O)_2SO_2}\ \xrightarrow[\mathrm{-Na_2SO_4}]{\mathrm{NaOH}}\ \mathsf{R\!\!-\!\!OCH_3}](40cb2412d3e10b1da4607b7acee13811.png)
![\mathsf{R\!\!-\!\!O\!\!-\!\!CH_2CH\!\!=\!\!CH_2}\ \xrightarrow[\mathrm{DMSO}]{\mathrm{(CH_3)_3COK}}\ \mathsf{R\!\!-\!\!O\!\!-\!\!CH\!\!=\!\!CH\!\!-\!\!CH_3}\ \xrightarrow[\mathrm{100\ ^\circ C}]{\mathrm{H^+}}\ \mathsf{ROH+CH_3CH_2CHO}](62b94e2504d21e128040d45eaf28fcee.png)

