- Острый миелоидный лейкоз
-
Острый миелоидный лейкоз 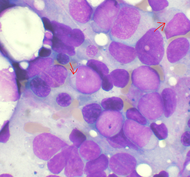
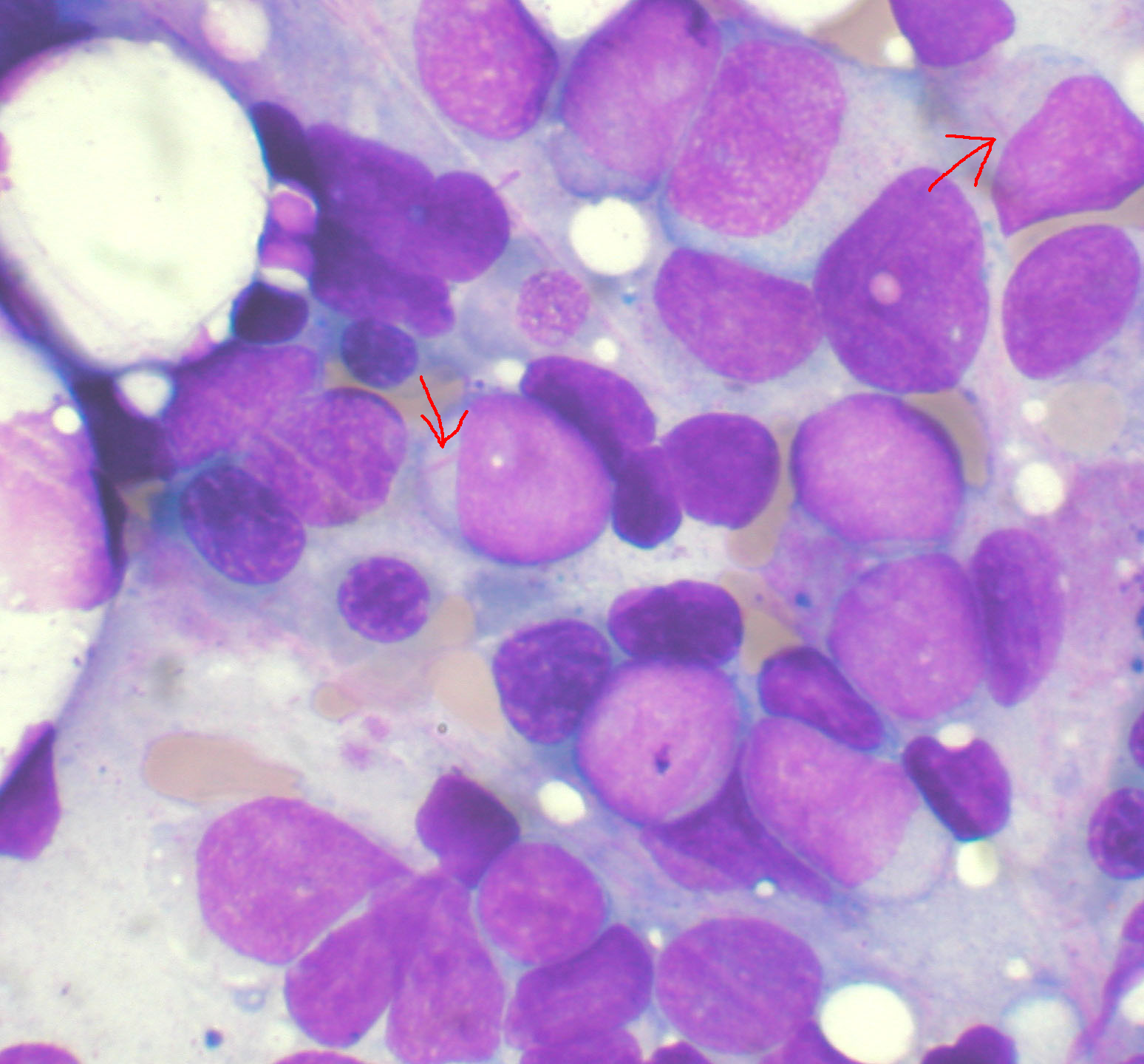
Мазок костного мозга при остром миелоидном лейкозе . Стрелками обозначены Тельца Ауэра. МКБ-10 C92.0 МКБ-9 205.0 МКБ-О M9861/3 OMIM 602439 DiseasesDB 203 eMedicine med/34 MeSH D015470 Острый миелоидный лейкоз (ОМЛ, острый нелимфобластный лейкоз, острый миелогенный лейкоз) — это злокачественная опухоль миелоидного ростка крови, при которой быстро размножаются изменённые белые кровяные клетки. Накапливаясь в костном мозге, они подавляют рост нормальных клеток крови. ОМЛ самый распространённый вид острого лейкоза у взрослых, заболеваемость им с возрастом увеличивается. Хотя острый миелоидный лейкоз заболевание относительно редкое — на его долю приходится лишь 1,2 % смертельных случаев злокачественных опухолей в США -[1], ожидается его учащение вместе с постарением населения.
Симптомы острого миелоидного лейкоза вызываются замещением нормального костного мозга лейкемическими клетками, что приводит к снижению количества красных кровяных клеток, тромбоцитов, и нормальных лейкоцитов. Болезнь проявляется быстрой утомляемостью, одышкой, частыми мелкими повреждениями кожи, повышенной кровоточивостью, частыми инфекционными поражениями. До сих пор явная причина заболевания неизвестна, однако некоторые факторы риска его возникновения выявлены. Как всякое острое заболевание, ОМЛ развивается быстро, и без лечения оборачивается летальным исходом за несколько месяцев, иногда — недель.
Встречаются несколько разновидностей ОМЛ, лечение и прогноз для них оказывается разным. Уровень выживаемости на протяжении пяти лет колеблется между 15-70 %, а частота ремиссии — от 78 до 33 % в зависимости от подвида заболевания. В начале ОМЛ лечат химиопрепаратами для того, чтобы добиться ремиссии; затем может проводиться поддерживающее химиолечение, или проводится пересадка кроветворных стволовых клеток. Последние исследования ОМЛ на генетическом уровне позволили разработать тесты, с помощью которых можно довольно точно определить вероятность выживания больного и эффективность того или иного лекарства для индивидуального случая ОМЛ.
Содержание
Классификация
Самые часто используемые схемы классификации ОМЛ — это давняя франко-американо-британская (ФАБ) система, и более современная система Всемирной организации здравоохранения (ВОЗ).
Классификация острого миелоидного лейкоза по системе Всемирной системы здравоохранения
Система классификации острого миелоидного лейкоза ВОЗ разработана с учётом системы ФАБ и имеет целью более эффективное клиническое применение и учитывает наиболее прогностически значимые признаки заболевания. Каждый из видов (категорий) ОМЛ по классификации ВОЗ включает в себя несколько подвидов (подкатегорий) описательного характера, представляющих интерес для гематологов и онкологов; однако, бо́льшая часть клинически важной информации в классификации ВОЗ взаимосвязана через распределение по перечисленным ниже подвидам.
Подвиды острого миелоидного лейкоза по классификации ВОЗ:[2]
Название подвида Описание МКБ-О ОМЛ с характерными генетическими изменениями Включает: - ОМЛ с транслокациями между хромосомой 8 и 21 [t(8;21)] (МКБ-О 9896/3); RUNX1/RUNX1T1
- ОМЛ инверсиями в хромосоме 16 [inv(16)] (МКБ-О 9871/3); CBFB/MYH11
- ОМЛ с транслокациями между хромосомой 15 и 17 [t(15;17)] (МКБ-О 9866/3); АРРК;ПМЛ — протеин
У больных с таким подвидом ОМЛ обычно высок уровень ремиссий и прогноз лучше сравнительно с ОМЛ других подвидов .
Несколько ОМЛ с дисплазией нескольких ростков Этот подвид включает больных с предшествующим миелодиспластическим синдромом (МДС) или миелопролиферативной болезнью (МПБ), которые переходят в ОМЛ. Этот подвид ОМЛ чаще встречается у пожилых людей и отличается неблагоприятным прогнозом . M9895/3 ОМЛ и МДС, связанные с предыдущим лечением Этот подвид ОМЛ включает больных, получавших химиолечение и/или лучевое лечение, после которых возник ОМЛ или МДС. При этих лейкозах могут быть характерные изменения в хромосомах, прогноз при них часто бывает хуже. . M9920/3 ОМД не подпадающие под признаки перечисленных подвидов Включает подвиды ОМЛ, не входящие в перечисленные выше. M9861/3 Бывают такие подвиды острого лейкоза, при которых изменённые лейкоциты невозможно определить как лимфоциты или гранулоциты, или когда присутствуют злокачественно изменённые клетки обоих ростков. Такие лейкозы иногда называют бифенотипными острыми лейкозами.
Франко-американско-британская классификация
Франко-американско-британская классификационная (ФАБ) система разделяет ОМЛ на 8 подвидов, от М0 по M7, основываясь на типах клеток — предшественниц лейкоцитов, и на степени зрелости изменённых клеток. Определение злокачественных клеток проводят на основании внешних признаков при световой микроскопии и/или цитогенетически, выявляя лежащие в основе отклонений изменения в хромосомах. У разных подвидов ОМЛ разные прогноз и ответ на лечение. Несмотря на преимущества классификации ВОЗ, система ФАБ до сих пор широко применяется. По ФАБ существует восемь подтипов ОМЛ.[1]
Подвид Название Цитогенетические изменения M0 минимально-дифференцированный острый миелобластный лейкоз M1 острый миелобластный лейкоз без созревания M2 острый миелобластный лейкоз с созреванием гранулоцитов t(8;21)(q22;q22), t(6;9) M3 промиелоцитарный, или острый промиелоцитарный лейкоз (ОПЛ) t(15;17) M4 острый миеломоноцитарный лейкоз inv(16)(p13q22), del(16q) M4eo миеломоноцитарный сочетанный с эозинофилией костного мозга inv(16), t(16;16) M5 острый монобластный лейкоз (M5a) или острый моноцитарный лейкоз (M5b) (M5b) del (11q), t(9;11), t(11;19) M6 острые эритроидные лейкозы, включая эритроцитарный лейкоз (M6a) и очень редкий чистый эритроидный лейкоз (M6b) M7 острый мегакариобластный лейкоз t(1;22) M8 острый базофильный лейкоз Необычные фенотипы острого миелоидного лейкоза
Морфологические подтипы ОМЛ включают многие исключительно редкие подтипы не включённые в классификацию ФАБ. Все они, за исключением острого миелоидного дендритноклеточного лейкоза включены в классификацию ВОЗ. В списке ниже перечислены эти подтипы.
- Острый базофильный лейкоз
- Острый эозинофильный лейкоз
- Тучноклеточный лейкоз
- Острый миелоидный дендритноклеточный лейкоз
- Острый панмиелоз с миелофиброзом
- Миелоидная саркома.
Проявления острого миелолейкоза
 Инфильтрированная лейкоцитами печень при миелолейкозе
Инфильтрированная лейкоцитами печень при миелолейкозе
Бо́льшая часть симптомов ОМЛ вызывается замещением нормальных клеток крови лейкозными клетками. Недостаточное образование лейкоцитов обусловливает высокую восприимчивость больного к инфекциям — несмотря на то, что лейкемические клетки происходят от предшественников лейкоцитов, способность противостоять инфектам у них отсутствует.[3] Снижение количества красных кровяных телец (анемия) может вызывать усталость, бледность, и одышку. Недостаток тромбоцитов может привести к легкой повреждаемости кожи и повышенной кровоточивости.
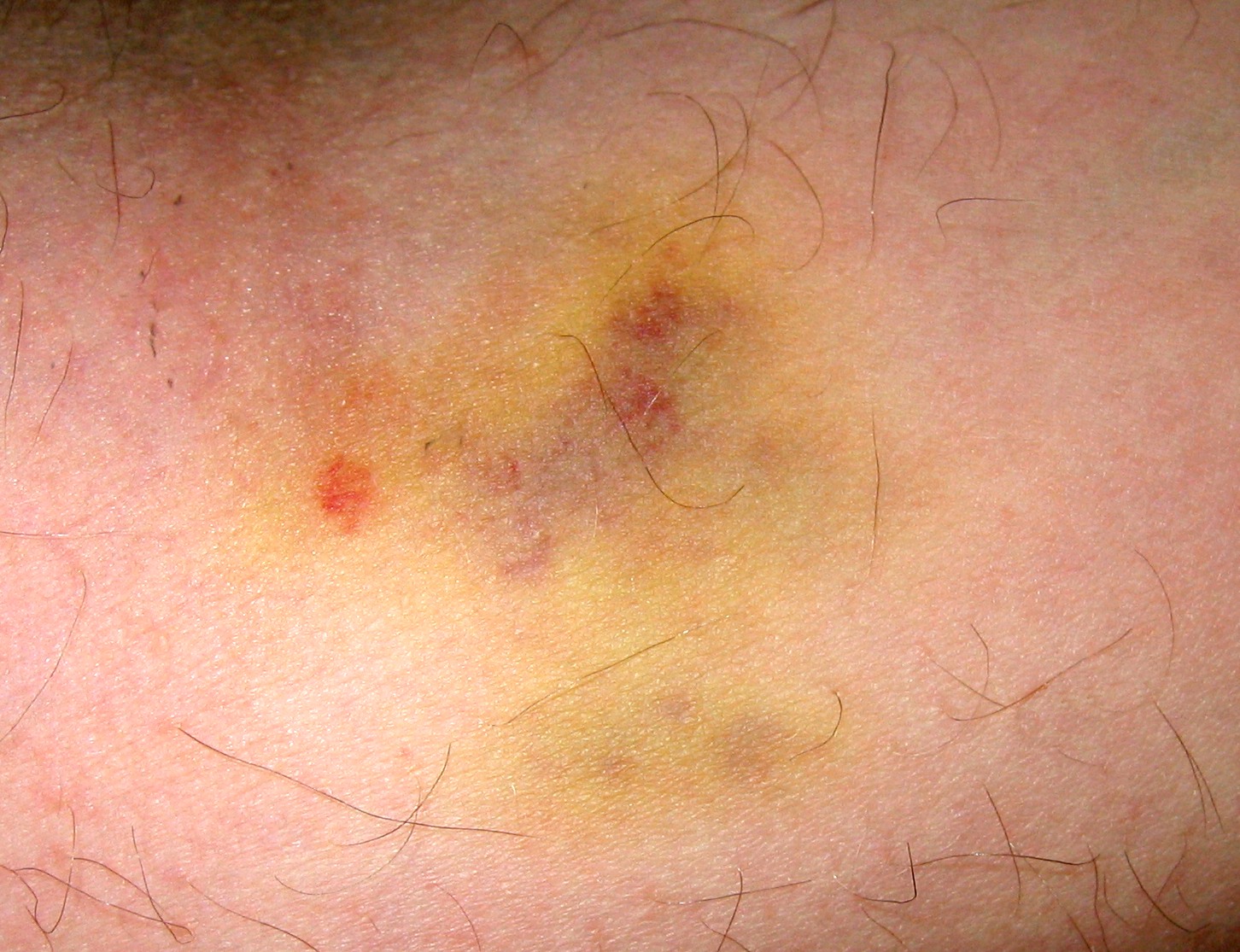
 Гематома.
Гематома.Ранние признаки ОМЛ часто неопределённы и неспецифичны, и могут походить на признаки гриппа или других распространённых болезней. Вот некоторые общие симптомы ОМЛ: лихорадка, усталость, потеря веса или снижение аппетита, одышка, анемия, повышенная повреждаемость кожи и слизистых оболочек и кровоточивость, петехии (плоские, размером с булавочную головку пятнышки внутри кожи на месте кровоизлияний), гематомы, боль в костях и суставах, и стойкие или частые инфекции.[3]
При ОМЛ может быть увеличение селезёнки но обычно оно незначительно и бессимптомно. Увеличение лимфоузлов при ОМЛ случается нечасто, в отличие от острого лимфобластного лейкоза. В 10 % случаев развиваются изменения кожи в виде кожного лейкоза. Изредка при ОМЛ возникает Синдром Свита, он же паранеопластический синдром — воспаление кожи вокруг поражённых хлоромой участков.[3]
У некоторых больных ОМЛ появляется припухлость дёсен из-за инфильтрации тканей лейкозными клетками. Изредка первым признаком лейкоза оказывается хлорома — плотная лейкемическая масса за пределами костного мозга. Иногда заболевание протекает бессимптомно, и лейкоз выявляется общим анализом крови в ходе профилактического осмотра.[4]
Причины
Был выявлен ряд факторов, способствующих возникновению ОМЛ — иные расстройства системы кроветворения, воздействие вредных веществ, ионизирующее излучение, и генетическое влияние.
Пре-лейкоз
"Пре-лейкозные нарушения кроветворения, такие, как миелодиспластический синдром или миелопролиферативный синдром могут привести к ОМЛ; вероятность заболевания зависит от формы миелодиспластического или миелопролиферативного синдрома.[5]
Воздействие химических веществ
Противоопухолевое химиотерапевтическое воздействие, особенно алкилирующими веществами, может увеличивать вероятность возникновения ОМЛ в последующем. Наивысшая вероятность заболевания приходится на 3—5 лет после химиотерапии.[6] Другие химиотерапевтические препараты особенно эпиподофилотоксины и антрациклины, также связываются с постхимиотерапевтическими лейкозами. лейкозы такого вида часто объясняют специфическими изменениями в хромосомах лейкозных клеток.[7]
Воздействие бензола и других ароматических органических растворителей, связанное с профессиональной деятельностью, в качестве возможной причины ОМЛ остаётся спорным. Бензол и многие его производные проявляют канцерогенные свойства in vitro. Данные некоторых наблюдений подтверждают возможность влияния профессиональных контактов с этими веществами на вероятность развития ОМЛ, однако другие исследования подтверждают, что если и существует такая опасность, то она является лишь добавочным фактором.[8][9]
Ионизирующее излучение
Воздействие ионизирующего излучения повышает вероятность заболеваня ОМЛ. У переживших атомную бомбардировку Хиросимы и Нагасаки заболеваемость ОМЛ повышена,[10] так же как у рентгенологов, получивших высокие дозы рентгеновского излучения в то время, когда меры радиологической защиты были недостаточными.[11]
Генетические факторы
Вероятно, существует наследственно повышенная вероятность заболевания ОМЛ. Есть большое количество сообщений о множестве семейных случаев ОМЛ, когда заболеваемость превышала среднестатистическую.[12][13][14][15] Вероятность возникновения ОМЛ у ближайших родственников больного втрое выше.[16]
Ряд врождённых состояний может повышать вероятность ОМЛ. Чаще всего это синдром Дауна, при котором вероятность ОМЛ повышена в 10 — 18 раз.[17]
Диагностика
Изменения соотношения клеточных элементов в общем анализе крови — первое, что наводит на мысль о возможности ОМЛ. Наиболее частым оказывается лейкоцитоз- повышение общего количества лейкоцитов, иногда с появлением бластных (незрелых) форм, но бывает так, что ОМЛ проявляется изолированным снижением тромбоцитов, эритроцитов, а количество лейкоцитов может оказаться даже сниженным (лейкопения).[18] Предварительный диагноз ОМЛ может быть определён в случае выявления в мазках периферической крови бластных форм лейкоцитов, но для окончательного диагноза должны быть выявлены соответствующие изменения в анализе аспирационного биоптата костного мозга.
Костный мозг и кровь исследуют световым микроскопированием и поточной цитометрией для установления диагноза и дифференцирования ОМЛ от других лейкозов, например, от острого лимфобластного лейкоза, а также для уточнения подтипа ОМЛ. Образец крови или костного мозга обычно проверяют на присутствие хромосомных транслокаций обычными цитогененитческими способами, или способом флуоресцентной гибридизации in situ. Генетические исследования проводят и для выявления характерных мутаций, которые могут влиять на исход заболевания — например, в FLT3, в нуклеоплазмине, или в KIT.[19]
Цитохимические красители для мазков крови и костного мозга оказываются очень полезными для дифференциальной диагностики ОМЛ и ОЛЛ, а также для выделения подтипов ОМЛ. Сочетание миелопероксидазы или красителя Суданский чёрный и неспцифического эстеразного красителя обеспечивает получение нужной информации в большинстве случаев. Реакции миелопероксидазы или Суданского чёрного оказываются самыми полезными для установления диагноза ОМЛ и его дифференцирования от ОЛЛ. Неспецифический краситель эстеразы используется для моноцитаронй составляющей острых миелолейкозов и для дифференцирования от незрелой формы монобластного лейкоза от ОЛЛ.[2]
Диагноз и классификация ОМЛ может оказаться непростой задачей, этим должен должен заниматься квалифицированный гематопатолог или гематолог. В явных случаях наличие некоторых характерный морфологических признаков, таких, как тельца Ауэра, или специфическия для АМЛ результаты поточной цитометрии дают возможность надёжно отделять ОМЛ от других лейкозов; однако при отсутствии такх явных признаков диагностика существенно осложняется.[20]
В соответствии с широко используемыми критериями классификации ВОЗ диагноз ОМЛ устанавливаетсяв том случае, если доказано если в крови и\или костном мозге более 20 % клеток представлены миелобластами.[21] ОМЛ следует тщательно дифференцировать от так называемых прелейкозных состояний, в частности, от миелодиспластического синдрома или миелопролиферативного синдрома, которые лечатся по разному.
Поскольку острый промиелоцитарный лейкоз (ОПЛ), требуя уникального способа лечения очень хорошо ему поддаётся, очень важно быстро подтвердить или отвергнуть этот подвид лейкоза. Для этого част о используют флюоресцентную гибридизацию in situ, выполненную на крови или костном мозге, поскольку ею легко выявляется хромосомная транслокация (t[15;17]) специфичная для ОПЛ.[22]
Патофизиология
Злокачественными клетками при ОМЛ выступают миелобасты. В нормальном гемопоэзе миелобласты являются незрелыми предшественниками лейкоцитов миелоидного ряда;нормальные миелобласты постепенно созревают, превращаясь в нормальные лейкоциты. Однако при ОМЛ в каком — то из миелобластов накапливаются генетические изменения, которые «замораживают» клетку в незрелом состоянии, останавливая процесс клеточной дифференциации. [23] Сама по себе такая мутация не вызывает лейкоз, но когда «останов дифференциации» сочетается с другими мутациями, которые приводят к утрате генетического контроля над ростом клеток, результатом оказывается неуправляемое размножение незрелого клеточного клона, определяющего клиническую сущность ОМЛ.[24]
Большое разнообразие и генетическая неоднородность ОМЛ происходит от того, что лейкозная трансформация может возникнуть на множестве этапов дифференциации клетки.[25] Современные схемы классификации ОМЛ признают факт зависимости свойств и поведения лейкозных клеток, а также течения лейкоза от того, на каком этапе остановилась дифференциация.
Специфические цитогенетическиеотклонения обнаруживаются у многих больных ОМЛ. Виды изменений хромосом часто прогностически значимы.[26] Хромосомные транслокации кодируют синтез гибридных белков, чаще всего, факторов транскрипции — вспомогательных белков — помощников РНК-полимераз, изменённые свойства которых могут привести к останову дифференциации.[27] Например, при остром промиелоцитарном лейкозе транслокация t(15; 17) вызывает синтез гибридного протеина PML-RARα, который связывается с рецептором ретиноевой кислоты в промоторах некоторых специфических для миелоидных клеток генов и останавливает в этих клетках дифференциацию.[28]
 Схема транслокации хромосом.
Схема транслокации хромосом.Клинические проявления и симптомы ОМЛ возникают потому что, умножаясь, клетки лейкозного клона мешают деятельности нормальных клеток и стремятся вытеснить их из костного мозга.[29] Это приводит к нейтропении, анемии, и тромбоцитопении. Чаще всего симптомы ОМЛ определяются недостаточностью нормальных клеток крови. В редких случаях у больных развиваются хлоромы — плотные опухоли из лейкозных клеток за прелами костного мозга, которые могут вызывать различные симптомы в зависимости от локализации хлоромы.[3]
Лечение
Лечение ОМЛ состоит в основном из химиотерапии, и делится на два этапа: индукция и постремиссионное лечение(или консолидация). Цель индукционной терапии является достижение полной ремиссии за счет уменьшения количества лейкозных клеток до не обнаруживаемого уровня; цель консолидирующей терапии заключается в ликвидации остаточных, не обнаруживаемых современными методами остатками болезни и излечение.[30]
Индукция
Для всех подтипов ОМЛ за исключением M3 по классификации ФАБ, обычно используют индукционную химиотерапию цитарабином и антрациклиновым антибиотиком (например, даунорубицином или идарубицином).[31] Этот способ индукционной химиотерапии известен под названием «7+3» — из-за того, что цитарабин вводят внутривенно капельно семь дней подряд, а затем три дня подряд (однократной внутривенной инъекцией) вводят антрациклиновый антибиотик. При таком способе лечения ремиссия наступает почти у 70 % больных ОМЛ.[32] Могут применяться и другие способы индукционного лечения, включая монотерапию высокими дозами цитарабина, или препаратами, находящимися на стадии исследования.[33][34] Вследствие токсического воздействия лечения, в том числе подавления миелоидного ростка и повышения вероятности инфекционных осложнений очень старым больным индукционная химиотерапия не предлагается, и назначается менее интенсивное паллиативное лечение химиопрепаратами. Подвид ОМЛ M3, также известный под названием острый промиелоцитарный лейкоз, почти во всех случаях лечится препаратом ПТРК (полностью транс- ретиноевая кислота) в дополнение к индукционной терапии.[35][36][37] При лечении острого промиелоцитарного лейкоза нужно учитывать возможность развития синдрома диссеминированного внутрисосудистого свёртывания вследствие поступления содержимого гранул промиелоцитов в периферическую кровь. Лечение острого промиелоцитарного лейкоза исключительно эффективно, это достоверно доказано множеством документированных случаев лечения.
Целью индукционного этапа лечения является достижение полной ремиссии. Полная ремиссия не означает, что заболевание полностью вылечено. Скорее, состояние полной ремиссии говорит о невозможности обнаружить болезнь существующими способами диагностики.[31] Полная ремиссия достигается у 50-70 % взрослых больных с впервые выявленным ОМЛ, разница зависит от прогностических факторов, о которых сказано выше.[38] Длительность ремиссии зависит от прогностических качеств исходного лейкоза. В основном, все случаи ремиссии без дополнительного, консолидирующего лечения закачиваются рецидивом.[39]
Консолидационное лечение
Даже после достижения полной ремиссии вероятно, немногие лейкозные клетки всё же выживают. Их так мало, что обнаружить их пока невозможно. В случае не проведения послеремиссионного, или консолидационного лечения почти у всех больных в конце — концов возникает рецидив.[40] Поэтому для того, чтобы избавиться от неопределимых больных клеток и предотвратить рецидив — то есть, достичь полного излечения, нужна дополнительная терапия. Вид лечения после достижения ремиссии определяется индивидуально в зависимости от прогностических факторов и общего состояния здоровья больного. При прогностически благоприятных подвидах лейкозов (например, при inv(16), t(8;21) и t(15;17) обычно назначают 3-5 дополнительных курса интенсивной химиотерапии, известной как консолидационное лечение. Больным с высоким риском рецидива (например, при наличии цитогенетических изменений, сопутствующего миелодиспластического синдрома, или при ОМЛ, связанном с предшествующим лечением обычно рекомендуется транплантация аллогенных стволовых клеток гемопоэтического ряда, если позволяет общее состояние и есть подходящий донор.[41][42] При ОМЛ со средней вероятностью рецидива (при нормальных цитогенетических показателях или с такими цитогенетическими изменениями, которые не попадают в группы риска)вопрос консолидационного лечения не столь ясен и определяется рядом специфических показателей — возрастом больного, общим состоянием его здоровья, системой ценностей, и наконец, наличием донора подходящих стволовых клеток.[42]
Тем больным, которым пересадка стволовых клеток после консолидационного лечения не показана, проводят иммунотерапию комбинацией гистамина гидрохлорида (цеплена) и пролейкина. Такое лечение позволяет снизить вероятность рецидива на 14 %, удлиняя ремиссию на 50 %.[43]
Таким образом, стандартной терапией ОМЛ признаны высокоинтенсивная химиотерапия (ВХТ) и трансплантация костного мозга. [44]
Однако результаты лечения, несмотря на относительно высокие ответы у молодых, остаются неудовлетворительными у лиц старше 65 лет (30-50%) связанные с ранней летальностью (10%) и непродолжительностью ремиссии. Больше половины с ОМЛ – это больные старшего возраста и/или со значимой сопутствующей патологией, которые, как правило, не могут получать высокотоксичные схемы химиотерапии, поэтому для их терапии применяют низкие дозы цитарабина цитарабина и поддерживающее лечение: антибиотики и гемотрансфузии.
С 2010 года в США для лечения ОМЛ рекомендовано применять гипометилирующие агенты (5-азацитидин, децитабин) у пациентов, которые не подходят для трансплантации костногомозговых клеток/интенсивной химиотерапии. [45] В процессе метилирования ДНК гипометилирующие агенты ковалентно связываются с ДНК-метилтрансферазой, что приводит к реактивации генов, после чего восстанавливается дифференцировка гемопоэтических клеток-предшественников и нормальное кроветворение. 5-азацитидин обладает двойным механизмом действия. Он встраивается не только в молекулу ДНК, но и в молекулу РНК. Тем самым 5-азацитидин понижает количество РНК в клетках, что приводит к цитостатическому эффекту вне зависимости от клеточной фазы.
На основании результатов исследования 3 фазы AZA-001 - международное, мультицентровое, контролируемое исследование в параллельных группах, в котором пациенты МДС высокого риска/ОМЛ (ВОЗ критерии) сравнивались со стандартно используемой терапией (сопроводительная терапия, интенсивная химиотерапия, низкие дозы цитарабина), азацитидин был зарегистрирован, в том числе и в РФ, для лечения этих групп больных. Было показано, что азацитидин в 2,5 раза увеличивает общую выживаемость пациентов с ОМЛ (критерии ВОЗ). [46]
Рецидив ОМЛ
При рецидиве ОМЛ единственным испытанным способом лечения, который может оказаться действенным, оказывается пересадка стволовых клеток при условии, если она ещё не применялась.[47][48][49]
При рецидиве ОМЛ больным, которым не планируется пересадка стволовых гемопоэтических клеток, или в случае рецидива после пересадки стволовых клеток можно предложить лечение в рамках клинического испытания, поскольку выбор среди обычных способов лечения весьма ограничен. В настоящее время проходит клинические испытания цитостатическое вещество клофарабин, также испытываются различные способы прицельной терапии с использованием ингибиторов фарнезилтрансферазы, децитабина и ингибиторов белка множественной лекарственной устойчивости, ингибиторы гистондеацетилазы цитарабина, блокаторы ангиогенеза, аналоги дезоксиаденозина. Иногда ограниченное количество методов терапии рецидивов ОМЛ вынуждает обращаться к паллиативному лечению.
После клинических испытаний триоксида мышьяка Управление по контролю за качеством пищевых продуктов и лекарственных препаратовСША одобрило его применение в качестве паллиативного средства лечения рецидивирующего острого промиелоцитарного лейкоза (ОПЛ). Также как и ПТРК триоксид мышьяка неэффективен при других подвидах ОМЛ.[50]
В 2000 году в США для лечения рецидивов ОМЛ у больных старше 60-ти лет, которых нельзя лечить высокими дозами химиопрепаратов, было разрешено применять гемтузумаб озогамицин (Милотарг), препарат, в котором, как предполагалось, цитотоксическое вещество — противоопухолевый антибиотик калихимицин — связан с моноклональным антителом, обеспечивающим доставку антибиотика точно в лейкозную клетку.[51] В настоящее время данный препарат снят с производства, т.к. в результате исследований было выявлено отсутствие разницы у пациентов принимавших и не принимавших милотарг.
Прогноз
 Хромосомная транслокация, (9;11), сочетающаяся с ОМЛ
Хромосомная транслокация, (9;11), сочетающаяся с ОМЛОстрый миелоидный лейкоз — излечимое заболевание. Вероятность выздоровления каждого пациента разная, поскольку определяется множеством прогностических факторов.[52]
Цитогенетика
Самым значимым для ОМЛ прогностическим фактором оказывается фактор цитогенетический, то есть «конструкция» хромосом лейкозных клеток. Некоторые цитогенетические отклонения связаны с весьма благоприятными исходами (например, транслокация 15;17 при остром промиелоцитарном лейкозе. Примерно у половины больных ОМЛ цитогенетическое состояние выглядит нормальным, таких пациентов относят к группе со средним вероятностью рецидива. Ряд цитогенетических изменений связан с неблагоприятным прогнозом и высокой вероятностью рецидива после лечения.[53][54][55]
Первая публикация, рассматривающая связь цитогенетических изменений с прогнозом появилась в отчёте Совета медицинских исследований в 1998 году.[56]
Исход Отклонения 5-летнее выживание Частота рецидивов Благоприятный t(8;21), t(15;17), inv(16) 70 % 33 % Удовлетворительный Не выявлено, +8, +21, +22, del(7q), del(9q), Нарушения 11q23, все остальные структурные или численные изменения 48 % 50 % Неблагоприятный −5, −7, del(5q), Нарушения 3q, Комбинированные цитогенетические нарушения 15 % 78 % Позже Юго-западная онкологическая группа и Восточная кооперативная онкологическая группа,[57] а затем и Группа Б по исследованию рака и лейкозов опубликовали другие, в основном совпадающие перечни прогностических цитогенетических изменений при лейкозах.[58]
Предшествующий миелодиспластический синдром и прогноз ОМЛ
У ОМЛ, развивающегося на фоне миелодиспластического синдрома или миелопролиферативного синдрома (так называемый вторичный ОМЛ) прогноз хуже, чем у ОМЛ, связанного с предшествующим лечением, который возникает после химиотерапии другого предшествующего злокачественного заболевания. Оба эти состояния связаны с высокой частотой неблагоприятных цитогенетических изменений.[59][60][61]
Другие прогностические признаки ОМЛ
Некоторые исследования связывают возраст после 60 лет и повышенный уровень лактатдегидрогеназы с повышенной вероятностью неблагоприятного исхода.[62] Как и при большинстве других злокачественных заболеваний общий соматический статус (то есть общее физическое состояние больного и его жизненная активность) тоже имеют большое прогностическое значение.
Доказано, что внутреннее тандемное удвоение FLT3-тирозинкиназы ухудшает прогноз ОМЛ.[63] Более агрессивное лечение таких больных, в частности, пересадкой стволовых клеток в первой ремиссии не увеличивало длительность выживания, поэтому в качестве благоприятного прогностического признака удвоение FLT3-тирозинкиназы определённого клинического значения не имеет.[64] Внутреннее тандемное удвоение FLT3-тирозинкиназы может быть связано с лейкостазом.[65]
Сейчас широко распространены исследования клинического значения мутаций гена CD117, ответственного за синтез рецептора фактора роста стволовых клеток c-KIT при ОМЛ. Важность этих исследований определяется появлением в клинической практике ингибиторов тирозинкиназы, таких, как иматиниб и сунитиниб, обладающих способностью останавливать действие рецептора c-KIT.[66]
Кроме этого, в качестве прогностических факторов и объектов лечебного воздействия изучаются гены CEBPA, BAALC, ERG и NPM1.
Общая оценка результатов лечения
Эффективность лечения в клинических испытаниях колеблется от 20 до 45 %.[67][68] Однако следует отметить, что в клинических испытаниях участвуют молодые больные, и также те, кто в состоянии перенести агрессивные методы лечения. Эффективность лечения для всех больных, включая престарелых и тех, кому агрессивное лечение противопоказано, скорее всего, ниже. В случае острого промиелоцитарного лейкоза эффективность лечения может достигать 98-ми%.[69]
Эпидемиология
Острый миелоидный лейкоз относительно редко встречающаеся злокачественное заболевание. Так, в США ежегодно выявляется 10 500 свежих случаев ОМЛ, а заболеваемость сохраняется неизменной с 1995 по 2005 г. Смертность от ОМЛ составляет 1,2 % всей онкологической смертности в США.[1]
Уровень заболеваемости ОМЛ увеличивается с возрастом, средний возраст выявления заболевания составляет 63 года. На ОМЛ приходится около 90 % всех острых лейкозов у взрослых, но у детей он встречается редко.[1]
 Заболеваемость ОМЛ в зависимости от возраста.
Заболеваемость ОМЛ в зависимости от возраста.Заболеваемость ОМЛ, связанным с предшествующим лечением (то-есть, ОМЛ, вызванным предшествующей химиотерапией) возрастает. В настоящее время такие формы достигают 10-20 % от всех случаев ОМЛ.[70] ОМЛ несколько чаще встречается у мужчин, заболеваемость соотносится как 1,3 к 1.[71]
Существуют некоторые географические отличия в заболеваемости ОМЛ. У взрослых высшая заболеваемость у взрослых приходится на Северную Америку, Европу и Океанию, а в Азии и Латинской Америке заболеваемость ОМЛ ниже.[72][73] И наоборот, детский ОМЛ в Северной Америке и в Индии встречается реже, чем других частях Азии.[74] Эти различия могут определяться генетическими особенностями населения и особенностями окружающей среды.
История
 Альфред Вельпо
Альфред ВельпоПервое описание лейкоза в медицинской литературе относится к 1827, когда французский врач Альфред-Арман-Луи-Мари Вельпо описал болезнь 63-х летнего садовника, которая проявилась лихорадкой, слабостью, камнями в почках и выраженным увеличением печени и селезёнки. Вельпо заметил, что кровь больного напоминала по консистенции «жидкую овсянку», и предположил, что это произошло из-за белых клеток крови.[75] В 1845 году работавший в Эдинбурге патолог Джон Хьюз Беннет опубликовал сообщение о нескольких случаях смерти больных, у которых оказалась увеличенной селезёнка, а «кровь была изменена по цвету и консистенции». Для описания изменений крови Беннет употребил термин «лейкоцитемия».[76]
Термин «лейкемия» был введён знаменитым немецким патологом Рудольфом Вирховом, в 1856 году. Будучи первым исследователем, применившим световой микроскоп в гистопатологии, Вирхов оказался первым учёным, описавшим избыточное количество белых кровяных клеток у больных с клиническими проявлениями, описанными Вельпо и Беннетом. Поскольку Вирхову была неизвестна причина избытка лейкоцитов, для определения состояния он употребил исключительно описательный термин «лейкемия» (Греческ: «белая кровь»).[77]
С развитием современных способов исследований пришло понимание многих деталей процесса возникновения и развития лейкозов. Уже в 1877 году Пауль Эрлих разработал способ окраски мазков крови, позволивший ему детально описать нормальные и изменённые лейкоциты. В 1889 году, чтобы разделить быстро развивающиеся смертельные и относительно медленно текущие хронические лейкозы Вильгельм Эбштайн ввёл термин «острый лейкоз». .[78] Термин «миелоидный» был введён Нойманом в 1869 году, когда он открыл, что лейкоциты образуются в костном мозге (древнегреческий: µυєλός, миелос = костный мозг), а не в селезёнке. Способ диагностики лейкоза исследованием мазка костного мозга впервые был описан Мозлером в 1879 году.[79] И, наконец, в 1900 году Негели, разделивший лейкозы на миелоцитарные и лимфоцитарные описал злокачественную клетку острого миелоидного лейкоза — миелобласт.[80][81]
 Миелобласты при ОМЛ M1 ФАБ
Миелобласты при ОМЛ M1 ФАБВ 2008 году была полностью определена последовательность генов в геноме больного ОМЛ. Геном при ОМЛ стал первым расшифрованным геномом при злокачественных опухолях. Полученная из лейкозных клеток ДНК была сопоставлена с ДНК, полученной из здоровой кожи[82],и в лейкозных клетках были выявлены мутации нескольких генов, ранее не считавшихся связанными с заболеванием.
См. также
- Острый лимфобластный лейкоз
- Хронический миелобластный лейкоз
- Хронический лимфолейкоз
- Хлорома
Ссылки
- Acute Myeloid Leukemia at American Cancer Society
- Acute Myeloid Leukemia at Leukemia & Lymphoma Society
- Childhood Acute Myeloid Leukemia at cchs.net
- PDQ statement on AML for health professionals at National Cancer Institute
Острый миелоидный лейкоз — одна из разновидностей лейкозов, характеризующаяся следующими признаками: острое (а иногда внезапное) начало болезни бурное (иногда молниеносное) и часто высокозлокачественное течение происхождение лейкозных клеток из миелоидного, а не лимфоидного ряда клеток костного мозга малое время удвоения общей массы бластных лейкозных клеток — иногда измеряемое сутками, чаще неделями, редко месяцами выраженная незрелость лейкозных клеток — более выраженная, чем при лимфоидных лейкозах без лечения всегда фатален нечувствительность или малая чувствительность лейкозных клеток к препаратам, специфичным для лимфоидных лейкозов, таким, как метотрексат или глюкокортикоиды
Примечания
- ↑ 1 2 3 Jemal A, Thomas A, Murray T, Thun M (2002). «Cancer statistics, 2002». CA Cancer J Clin 52 (1): 23–47. DOI:10.3322/canjclin.52.1.23. PMID 11814064.
- ↑ 1 2 Vardiman JW, Harris NL, Brunning RD (2002). «The World Health Organization (WHO) classification of the myeloid neoplasms». Blood 100 (7): 2292–302. DOI:10.1182/blood-2002-04-1199. PMID 12239137.
- ↑ 1 2 3 4 Hoffman Ronald et al.' Hematology: Basic Principles and Practice. — 4th.. — St. Louis, Mo.: Elsevier Churchill Livingstone, 2005. — P. 1074–75. — ISBN 0-443-06629-9
- ↑ Abeloff Martin et al.' Clinical Oncology. — 3rd.. — St. Louis, Mo.: Elsevier Churchill Livingstone, 2004. — P. 2834. — ISBN 0-443-06629-9
- ↑ Sanz G, Sanz M, Vallespí T, Cañizo M, Torrabadella M, García S, Irriguible D, San Miguel J (1989). «Two regression models and a scoring system for predicting survival and planning treatment in myelodysplastic syndromes: a multivariate analysis of prognostic factors in 370 patients.». Blood 74 (1): 395–408. PMID 2752119.
- ↑ Le Beau M, Albain K, Larson R, Vardiman J, Davis E, Blough R, Golomb H, Rowley J (1986). «Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7». J Clin Oncol 4 (3): 325–45. PMID 3950675.
- ↑ Thirman M, Gill H, Burnett R, Mbangkollo D, McCabe N, Kobayashi H, Ziemin-van der Poel S, Kaneko Y, Morgan R, Sandberg A (1993). «Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations». N Engl J Med 329 (13): 909–14. DOI:10.1056/NEJM199309233291302. PMID 8361504.
- ↑ Austin H, Delzell E, Cole P (1988). «Benzene and leukemia. A review of the literature and a risk assessment.». Am J Epidemiol 127 (3): 419–39. PMID 3277397.
- ↑ Linet, MS. The Leukemias: Epidemiologic Aspects. Oxford University Press, New York 1985.
- ↑ Bizzozero O, Johnson K, Ciocco A (1966). «Radiation-related leukemia in Hiroshima and Nagasaki, 1946–1964. I. Distribution, incidence and appearance time». N Engl J Med 274 (20): 1095–101. PMID 5932020.
- ↑ Yoshinaga S, Mabuchi K, Sigurdson A, Doody M, Ron E (2004). «Cancer risks among radiologists and radiologic technologists: review of epidemiologic studies». Radiology 233 (2): 313–21. DOI:10.1148/radiol.2332031119. PMID 15375227.
- ↑ Taylor GM, Birch JM The hereditary basis of human leukemia // Leukemia / Henderson ES, Lister TA, Greaves MF. — 6th. — Philadelphia: WB Saunders, 1996. — P. 210. — ISBN 0-7216-5381-2
- ↑ Horwitz M, Goode EL, Jarvik GP (1996). «Anticipation in familial leukemia». Am. J. Hum. Genet. 59 (5): 990–8. PMID 8900225. Полный текст в свободном доступе на сайте PMC: 1914843
- ↑ Crittenden LB (1961). «An interpretation of familial aggregation based on multiple genetic and environmental factors». Ann. N. Y. Acad. Sci. 91: 769–80. DOI:10.1111/j.1749-6632.1961.tb31106.x. PMID 13696504.
- ↑ Horwitz M (1997). «The genetics of familial leukemia». Leukemia 11 (8): 1347–59. DOI:10.1038/sj.leu.2400707. PMID 9264391.
- ↑ Gunz FW, Veale AM (1969). «Leukemia in close relatives--accident or predisposition?». J. Natl. Cancer Inst. 42 (3): 517–24. PMID 4180615.
- ↑ Evans D, Steward J (1972). «Down's syndrome and leukaemia». Lancet 2 (7790): 1322. DOI:10.1016/S0140-6736(72)92704-3. PMID 4117858.
- ↑ Abeloff, Martin et al. (2004), p. 2834.
- ↑ Baldus CD, Mrózek K, Marcucci G, Bloomfield CD (June 2007). «Clinical outcome of de novo acute myeloid leukaemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review». Br. J. Haematol. 137 (5): 387–400. DOI:10.1111/j.1365-2141.2007.06566.x. PMID 17488484.
- ↑ Abeloff, Martin et al. (2004), p. 2835.
- ↑ Harris N, Jaffe E, Diebold J, Flandrin G, Muller-Hermelink H, Vardiman J, Lister T, Bloomfield C (1999). «The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997». Ann Oncol 10 (12): 1419–32. DOI:10.1023/A:1008375931236. PMID 10643532.
- ↑ Grimwade D, Howe K, Langabeer S, Davies L, Oliver F, Walker H, Swirsky D, Wheatley K, Goldstone A, Burnett A, Solomon E (1996). «Establishing the presence of the t(15;17) in suspected acute promyelocytic leukaemia: cytogenetic, molecular and PML immunofluorescence assessment of patients entered into the M.R.C. ATRA trial. M.R.C. Adult Leukaemia Working Party.». Br J Haematol 94 (3): 557–73. PMID 8790159.
- ↑ Fialkow PJ (1976). «Clonal origin of human tumors». Biochim. Biophys. Acta 458 (3): 283–321. DOI:10.1016/0304-419X(76)90003-2. PMID 1067873.
- ↑ Fialkow PJ, Janssen JW, Bartram CR (Apr 1991). «Clonal remissions in acute nonlymphocytic leukemia: evidence for a multistep pathogenesis of the malignancy» (PDF). Blood 77 (7): 1415–7. PMID 2009365.
- ↑ Bonnet D, Dick JE (1997). «Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell». Nat. Med. 3 (7): 730–7. DOI:10.1038/nm0797-730. PMID 9212098.
- ↑ Abeloff, Martin et al. (2004), pp. е2831-32.
- ↑ Wintrobe's Clinical Hematology / Greer JP et al.. — 11th. — Philadelphia: Lippincott, Williams, and Wilkins, 2004. — P. 2045–2062. — ISBN 0781736501
- ↑ Melnick A, Licht JD (May 1999). «Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia». Blood 93 (10): 3167–215. PMID 10233871.
- ↑ Abeloff, Martin et al. (2004), p. 2828.
- ↑ Acute myeloid leukemia at Mount Sinai Hospital
- ↑ 1 2 Abeloff, Martin et al. (2004), pp. 2835-39.
- ↑ Bishop J (1997). «The treatment of adult acute myeloid leukemia». Semin Oncol 24 (1): 57–69. PMID 9045305.
- ↑ Weick JK, Kopecky KJ, Appelbaum FR, et al. (Oct 1996). «A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: a Southwest Oncology Group study» (PDF). Blood 88 (8): 2841–51. PMID 8874180.
- ↑ Bishop JF, Matthews JP, Young GA, et al. (Mar 1996). «A randomized study of high-dose cytarabine in induction in acute myeloid leukemia» (PDF). Blood 87 (5): 1710–7. PMID 8634416.
- ↑ Huang ME, Ye YC, Chen SR, et al. (Aug 1988). «Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia» (PDF). Blood 72 (2): 567–72. PMID 3165295.
- ↑ Tallman MS, Andersen JW, Schiffer CA, et al. (1997). «All-trans-retinoic acid in acute promyelocytic leukemia». N. Engl. J. Med. 337 (15): 1021–8. DOI:10.1056/NEJM199710093371501. PMID 9321529.
- ↑ Fenaux P, Chastang C, Chevret S, et al. (Aug 1999). «A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group». Blood 94 (4): 1192–200. PMID 10438706.
- ↑ Estey E (2002). «Treatment of acute myelogenous leukemia». Oncology (Williston Park) 16 (3): 343–52, 355–6; discussion 357, 362, 365–6. PMID 15046392.
- ↑ Cassileth P, Harrington D, Hines J, Oken M, Mazza J, McGlave P, Bennett J, O'Connell M (1988). «Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia». J Clin Oncol 6 (4): 583–7. PMID 3282032.
- ↑ Cassileth PA, Harrington DP, Hines JD, et al. (1988). «Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia». J. Clin. Oncol. 6 (4): 583–7. PMID 3282032.
- ↑ Mayer RJ, Davis RB, Schiffer CA, et al. (1994). «Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B». N. Engl. J. Med. 331 (14): 896–903. DOI:10.1056/NEJM199410063311402. PMID 8078551.
- ↑ 1 2 Appelbaum FR, Baer MR, Carabasi MH, et al. (2000). «NCCN Practice Guidelines for Acute Myelogenous Leukemia». Oncology (Williston Park, N.Y.) 14 (11A): 53–61. PMID 11195419.
- ↑ Brune M, Castaigne S, Catalano J, et al. (July 2006). «Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial». Blood 108 (1): 88–96. DOI:10.1182/blood-2005-10-4073. PMID 16556892.
- ↑ Савченко В.Г., Паровичникова Е.Н.,et al. (2004). «Лечение острых лейкозов.». М.:Медпресс-информ 224.
- ↑ NCCN - Evidence-Based Cancer Guidelines, Oncology Drug Compendium, Oncology Continuing Medical Education
- ↑ Fenaux P., Mufti J., Hellstrom-Lindberg E., et al. (2010). «Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia». J Clin Oncol 28(4): 562-569.
- ↑ Abeloff, Martin et al. (2004), pp. 2840-41.
- ↑ Appelbaum FR (2001). «Editorial: Кому же при ОМЛ можно пересаживать стволовые клетки?». Leukemia 15 (4): 680–2. PMID 11368380.
- ↑ Appelbaum FR (2002). «Keynote address: hematopoietic cell transplantation beyond first remission». Leukemia 16 (2): 157–9. DOI:10.1038/sj.leu.2402345. PMID 11840278.
- ↑ Soignet SL, Frankel SR, Douer D, et al. (Sep 2001). «United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia». J. Clin. Oncol. 19 (18): 3852–60. PMID 11559723.
- ↑ Sievers EL, Larson RA, Stadtmauer EA, et al. (Jul 2001). «Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse». J. Clin. Oncol. 19 (13): 3244–54. PMID 11432892.
- ↑ Estey E (2001). «Prognostic factors in acute myelogenous leukemia». Leukemia 15 (4): 670–2. PMID 11368376.
- ↑ Wheatley K, Burnett A, Goldstone A, Gray R, Hann I, Harrison C, Rees J, Stevens R, Walker H (1999). «A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council's Adult and Childhood Leukaemia Working Parties.». Br J Haematol 107 (1): 69–79. PMID 10520026.
- ↑ Slovak M, Kopecky K, Cassileth P, Harrington D, Theil K, Mohamed A, Paietta E, Willman C, Head D, Rowe J, Forman S, Appelbaum F (15 December 2000). «Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study.». Blood 96 (13): 4075–83. PMID 11110676.
- ↑ Byrd J, Mrózek K, Dodge R, Carroll A, Edwards C, Arthur D, Pettenati M, Patil S, Rao K, Watson M, Koduru P, Moore J, Stone R, Mayer R, Feldman E, Davey F, Schiffer C, Larson R, Bloomfield C (2002). «Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461).». Blood 100 (13): 4325–36. DOI:10.1182/blood-2002-03-0772. PMID 12393746.
- ↑ Grimwade D, Walker H, Oliver F, et al. (1 October 1998). «The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties». Blood 92 (7): 2322–33. PMID 9746770.
- ↑ Slovak ML, Kopecky KJ, Cassileth PA, et al. (2000). «Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study». Blood 96 (13): 4075–83. PMID 11110676.
- ↑ Byrd J, Mrózek K, Dodge R, Carroll A, Edwards C, Arthur D, Pettenati M, Patil S, Rao K, Watson M, Koduru P, Moore J, Stone R, Mayer R, Feldman E, Davey F, Schiffer C, Larson R, Bloomfield C (2002). «Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461).». Blood 100 (13): 4325–36. DOI:10.1182/blood-2002-03-0772. PMID 12393746.
- ↑ Thirman M, Larson R (1996). «Therapy-related myeloid leukemia.». Hematol Oncol Clin North Am 10 (2): 293–320. DOI:10.1016/S0889-8588(05)70340-3. PMID 8707757.
- ↑ Rowley J, Golomb H, Vardiman J (1 October 1981). «Nonrandom chromosome abnormalities in acute leukemia and dysmyelopoietic syndromes in patients with previously treated malignant disease.». Blood 58 (4): 759–67. PMID 7272506.
- ↑ Pedersen-Bjergaard J, Andersen M, Christiansen D, Nerlov C (2002). «Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia.». Blood 99 (6): 1909–12. DOI:10.1182/blood.V99.6.1909. PMID 11877259.
- ↑ Haferlach T, Schoch C, Löffler H, et al. (2003). «Morphologic dysplasia in de novo acute myeloid leukemia (AML) is related to unfavorable cytogenetics but has no independent prognostic relevance under the conditions of intensive induction therapy: results of a multiparameter analysis from the German AML Cooperative Group studies». J. Clin. Oncol. 21 (2): 256–65. DOI:10.1200/JCO.2003.08.005. PMID 12525517.
- ↑ Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Löffler H, Sauerland C, Serve H, Büchner T, Haferlach T, Hiddemann W (2002). «Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease». Blood 100 (1): 59–66. DOI:10.1182/blood.V100.1.59. PMID 12070009.
- ↑ Gale RE, Hills R, Kottaridis PD, et al. (2005). «No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML10 and 12 trials». Blood 106 (10): 3658–65. DOI:10.1182/blood-2005-03-1323. PMID 16076872.
- ↑ Thornton KA, Levis M (2007). «Images in clinical medicine. FLT3 Mutation and acute myelogenous leukemia with leukostasis». N. Engl. J. Med. 357 (16): 1639. DOI:10.1056/NEJMicm064764. PMID 17942876.
- ↑ Paschka P, Marcucci G, Ruppert AS, et al. (2006). «Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study». J. Clin. Oncol. 24 (24): 3904–11. DOI:10.1200/JCO.2006.06.9500. PMID 16921041.
- ↑ Cassileth PA, Harrington DP, Appelbaum FR, et al. (1998). «Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission». N. Engl. J. Med. 339 (23): 1649–56. DOI:10.1056/NEJM199812033392301. PMID 9834301.
- ↑ Matthews JP, Bishop JF, Young GA, et al. (2001). «Patterns of failure with increasing intensification of induction chemotherapy for acute myeloid leukaemia». Br. J. Haematol. 113 (3): 727–36. DOI:10.1046/j.1365-2141.2001.02756.x. PMID 11380464.
- ↑ Sanz MA, Lo Coco F, Martín G, et al. (15 August 2000). «Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups». Blood 96 (4): 1247–53. PMID 10942364.
- ↑ Leone G, Mele L, Pulsoni A, Equitani F, Pagano L (1 October 1999). «The incidence of secondary leukemias». Haematologica 84 (10): 937–45. PMID 10509043.
- ↑ Greenlee RT, Hill-Harmon MB, Murray T, Thun M (2001). «Cancer statistics, 2001». CA Cancer J Clin 51 (1): 15–36. DOI:10.3322/canjclin.51.1.15. PMID 11577478.
- ↑ Linet MS The leukemias: Epidemiologic aspects. // Monographs in Epidemiology and Biostatistics / Lilienfeld AM. — New York: Oxford University Press, 1985. — P. I. — ISBN 0195034481
- ↑ Aoki K, Kurihars M, Hayakawa N, et al. Death Rates for Malignant Neoplasms for Selected Sites by Sex and Five-Year Age Group in 33 Countries 1953–57 to 1983–87. — Nagoya, Japan: University of Nagoya Press, International Union Against Cancer, 1992.
- ↑ Bhatia S, Neglia JP (1995). «Epidemiology of childhood acute myelogenous leukemia». J. Pediatr. Hematol. Oncol. 17 (2): 94–100. DOI:10.1097/00043426-199505000-00002. PMID 7749772.
- ↑ Hoffman et al. 2005, pg 1071
- ↑ Bennett JH (1845). «Two cases of hypertrophy of the spleen and liver, in which death took place from suppuration of blood». Edinburgh Med Surg J 64: 413.
- ↑ Virchow R Die Leukämie // Gesammelte Abhandlungen zur Wissenschaftlichen Medizin / Virchow R. — Frankfurt: Meidinger, 1856. — P. 190.
- ↑ Ebstein W (1889). «Über die acute Leukämie und Pseudoleukämie». Deutsch Arch Klin Med 44: 343.
- ↑ Mosler F (1876). «Klinische Symptome und Therapie der medullären Leukämie». Berl Klin Wochenschr 13: 702.
- ↑ Naegeli O (1900). «Über rothes Knochenmark und Myeloblasten». Deutsch Med Wochenschr 26: 287.
- ↑ Zhen-yi, Wang (2003). «Ham-Wasserman Lecture: Treatment of Acute Leukemia by Inducing Differentiation and Apoptosis». Hematology 2003: 1. DOI:10.1182/asheducation-2003.1.1. PMID 14633774.
- ↑ Ley TJ, Mardis ER, Ding L, et al. (2008). «DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome». Nature 456 (7218): 66–72. DOI:10.1038/nature07485. PMID 18987736.

Кровь Кроветворение Компоненты Плазма • Эритроциты • Гематокрит • Тромбоциты • Лейкоциты (Гранулоциты (Нейтрофилы • Эозинофилы • Базофилы) • Агранулоциты (Лимфоциты (T- • B- • NK-) • Моноцит))
Биохимия Группа крови • Резус-фактор • Буферные системы (Ацидоз • Алкалоз) • Сыворотка • Гликемия (Гипер- • Гипо-)
Заболевания См. также Опухоли и онкология Доброкачественные опухоли • Предраки • Рак in situ • Злокачественные опухоли • Промежуточные опухоли Топография Голова и шея • ЦНС • Головной мозг • Глаза • Полость рта • Гортань • Щитовидная железа • Пищевод • Желудок • Двенадцатиперстная кишка • Печень • Желчный пузырь • Поджелудочная железа • Толстая кишка • Прямая кишка • Анус • Лёгкие • Средостение • Почки • Мочевой пузырь • Эндометрий • Шейка матки • Яичники • Молочная железа • Простата • Яички • Половой член • Кожа • Кости • APUD-система • островки Лангерганса
Морфология Эпителий
и железыпапиллома • аденома, фиброаденома, цистаденома, аденоматозный полип • неинвазивная карцинома • базалиома • плоскоклеточный рак • аденокарцинома • коллоидный рак • солидный рак • мелкоклеточный рак • фиброзный рак • медуллярный рак • саркома • карцинома
Мезенхима фиброма (десмоид) • гистиоцитома • липома • гибернома • лейомиома • рабдомиома • зернисто-клеточная опухоль • гемангиома • гломусная опухоль • лимфангиома • синовиома • мезотелиома • остеобластома • хондрома • хондробластома • гигантоклеточная опухоль • фибросаркома • липосаркома • лейомиосаркома • рабдомиосаркома • ангиосаркома • лимфангиосаркома • остеогенная саркома • хондросаркома
Меланинобразующая
тканьНервная система
и оболочки мозгаастроцитома • астробластома •
 гемангиобластома • олигодендроглиома • олигодендроглиобластома • пинеалома •
гемангиобластома • олигодендроглиома • олигодендроглиобластома • пинеалома •  эпендимома • эпендимобластома • опухоли сосудистого сплетения ( хориоидпапиллома • хориоидкарцинома) • ганглионеврома • ганглионейробластома • нейробластома • медуллобластома • глиобластома • менингиома • менингиальная саркома • симпатобластома • ганглионейробластома • хемодектома • невринома ( Невринома слухового нерва) • нейрофиброматоз ( Нейрофиброматоз I типа • Нейрофиброматоз II типа) • нейрогенная саркома • краниофарингиома
эпендимома • эпендимобластома • опухоли сосудистого сплетения ( хориоидпапиллома • хориоидкарцинома) • ганглионеврома • ганглионейробластома • нейробластома • медуллобластома • глиобластома • менингиома • менингиальная саркома • симпатобластома • ганглионейробластома • хемодектома • невринома ( Невринома слухового нерва) • нейрофиброматоз ( Нейрофиброматоз I типа • Нейрофиброматоз II типа) • нейрогенная саркома • краниофарингиомаСистема крови Тератомы Лечение Родственные структуры Киста • Дисплазия • Гамартома • Узел • Полип • Псевдокиста
Прочее Гены опухолевой супрессии • Онкоген • Стадирование • Градации • Канцерогенез • Метастазирование • Канцероген • Исследования • Паранеопластические феномены • МКБ-О • Список онкологических терминов
Категории:- Заболевания по алфавиту
- Лейкозы




Wikimedia Foundation. 2010.