- Получение спиртов
-
Спирты являются обширным и очень разнообразным классом органических соединений: они широко распространены в природе, имеют важнейшее промышленное значение и обладают исключительными химическими свойствами.
Существует огромное количество методов получения спиртов, при этом их можно разбить на две условных группы:
- химические способы получения спиртов — синтетические спирты;
- биохимические способы получения спиртов — биоспирты.
Химические способы получения спиртов
Занимая одну из центральных позиций в органической химии, спирты могут быть получены из множества других соединений. На практике в качестве исходных веществ для синтеза спиртов наиболее часто используют [1][2] :
- алкилгалогениды — щелочной гидролиз или реакция с супероксидом калия;
- алкены — кислотная гидратация, реакция гидроксимеркурирования-демеркурирования или гидроборирование с последующим окислением, а также промышленные методы оксо-синтеза;
- карбонильные соединения — восстановление или взаимодействие с реактивами Гриньяра.
Далее в разделе будет подробно рассмотрена химия существующих методов получения одноатомных спиртов. Промышленные аспекты получения спиртов, включая биохимические методы синтеза, подробно рассмотрены в подразделе «Получение спиртов в промышленности».
Краткий обзор методов органического синтеза многоатомных спиртов будет рассмотрен в соответствующем подразделе.
Получение спиртов из галогеноуглеводородов
Галогенпроизводные углеводородов под действием оснований трансформируются с образованием спиртов (реакция нуклеофильного замещения).
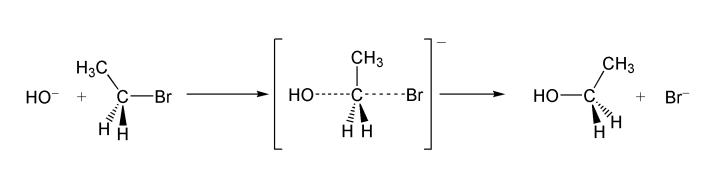
Обычно, первичные и вторичные галогенуглеводороды вступают в реакцию по одностадийному SN2 механизму[3]. Пример — гидролиз бромэтана:
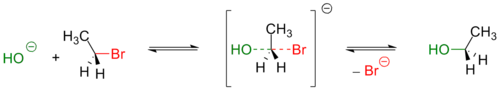
Реакции такого типа, обычно, происходят стереоспецифично — с обращением геометрической конфигурации исходного вещества[3]. Реакционная способность алкилгалогенидов уменьшается при переходе от производных йода к производным фтора [4] При этом фторпроизводные устойчивы к нуклеофильному замещению в обычных условиях и практически не используются для получения спиртов.
Первичные хлоралканы удовлетворительно гидролизуются под действием водного раствора щёлочи при нагревании[5]:

Для реакций, протекающих по SN2 механизму, используют только полярные растворители, причем скорость превращения возрастает при использовании вместо протонных растворителей (например: вода или спирт) апротонные (например: диметилсульфоксид); при этом в апротонных растворителях нуклеофильность уходящих групп будет иной[3]:

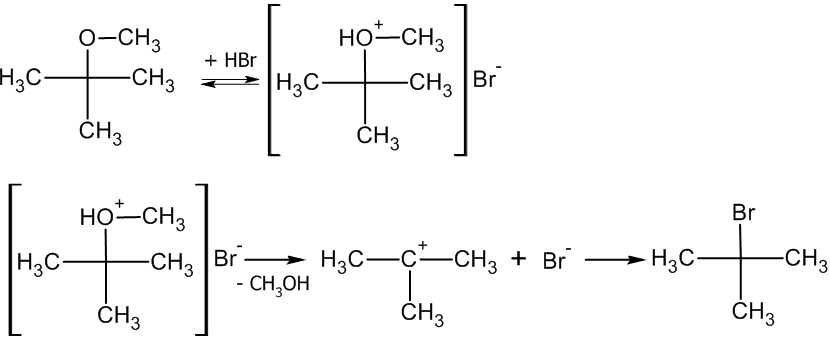
Третичные и в меньшей степени вторичные галогенуглеводороды гидролизуются по двухстадийному SN1 механизму[3]:
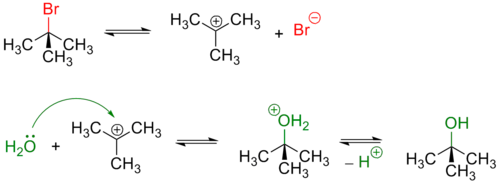
Реакция, протекающая по SN1 механизму проводят в полярных протонных растворителях, чаще всего воде или водном растворе метилового или этилового спирта.
Из-за устойчивости карбкатиона по такому механизму гидролизуются галогеналкены:

Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учета фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта.
Для высокореакционных реагентов используют мягкое замещение с использованием соединений одновалентного серебра или двухвалентной ртути[5]:
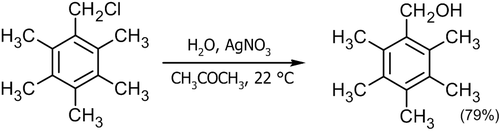
![\mathsf{C_6H_{13}\!\!-\!\!CHBr\!\!-\!\!CH_3+H_2O}\ \xrightarrow[25^oC,\ CH_3OC_2H_4OCH_3]{Hg(ClO_4)_2}\ \mathsf{C_6H_{13}\!\!-\!\!CH(OH)\!\!-\!\!CH_3\ _{(88%)}+HBr}](15a695ae530495b8f9afa1190ea635aa.png)
В современной лабораторной практике описанные выше реакции сольволиза проводят достаточно редко, так как спирты — более доступные полупродукты — являются исходным объектом для синтеза галогенпроизводных. Кроме того, следует помнить, что помимо изменения стереохимии исходных компонентов, реакции замещения конкурируют с элиминированием, а также перегруппировками, что часто приводит к нежелательным продуктам[3]:


В то же время существует достаточно новый метод превращения в спирты алкилгалогенидов действием на последние супероксида калия в среде диметилсульфоксида в присутствии 18-краун-6 полиэфира, при этом происходит практически полное геометрическое обращение[2]:
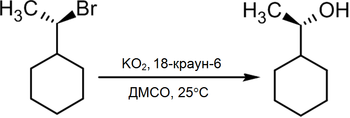
Получение спиртов из алкенов
Гидратация алкенов
Кислотная гидратация алкенов историческим была первым синтетическим методом получения спиртов (см. подраздел «История открытия спиртов»).
Общий механизм процесса (реакция электрофильного присоединения AdE2) представлен ниже[6]:

![\mathsf{R\!\!-\!\!CH\!\!=\!\!CH_2+H_3O^+}\rightleftarrows\mathsf{[R\!\!-\!\!CH^+\!\!-\!\!CH_3]+H_2O}\rightarrow\mathsf{R\!\!-\!\!CH(OH)\!\!-\!\!CH_3+H^+}](e703e803e92d4c9a4602359891cd0cb4.png)
Присоединение происходит по правилу Марковникова.
В случае использования серной кислоты в качестве катализатора промежуточным продуктом является эфир серной кислоты (R-CH(OSO2OH)-CH3), который в условиях реакции полностью гидролизуется до спирта[6].
Для проведения реакции кроме серной кислоты используют и другие реагенты: смесь муравьиной и каталитического количества серной кислоты (в отдельных случаях позволяет добиться стереоспецифичности), смесь муравьиной и хлорной кислоты, трифторуксуную кислоту и др[7].
Реакции вторичных алкенов, вследствие перегруппировок карбокатионов, часто приводят к образованию смеси продуктов, что затрудняет их использование для получения вторичных спиртов[8]:
![\mathsf{CH_3(CH_2)_3CH\!\!=\!\!CH_2+H_2O}\ \xrightarrow[85-100^oC]{HCOOH,\ HClO_4}\ \ \mathsf{CH_3(CH_2)_3CH(OH)CH_3+CH_3(CH_2)_2CH(OH)CH_2CH_3}](058656981ce792a6ff77076a24896eb0.png)
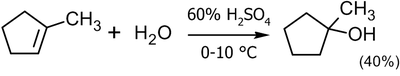
В лабораторной практике метод кислотной гидратации применим весьма ограниченно как из-за перспективы получения смеси продуктов, так и низких выходов. Чаще его используют для получения третичных спиртов, но и в этом случае выход, обычно, не превышает 40-45 %[8]:
![\mathsf{(CH_2)_2C\!\!=\!\!CH_2+H_2O}\ \xrightarrow[10-20^oC]{65% H_2SO_4}\ \ \mathsf{(CH_3)_2C(OH)CH_3}\ _{(45%)}](55a691da07be8b86ce10f5bf9762109c.png)
В промышленности, помимо жидкофазной используют прямую газофазную гидратацию алкенов. В качестве катализаторов используется фосфорная кислота на твердом носителе при 200—300 °C и давлении 2-8 МПа; при этом выход спиртов достигает 95 %[9]:
![\mathsf{CH_2\!\!=\!\!CH_2+H_2O}\ \xrightarrow[300^oC,\ 70-80\ ATM]{H_3PO_4/SiO_2}\ \ \mathsf{CH_3CH_2OH}](743a5aed9e36d2d7da3a987734dc88c5.png)
![\mathsf{CH_3CH\!\!=\!\!CH_2+H_2O}\ \xrightarrow[200^oC,\ 20-30\ ATM]{H_3PO_4/SiO_2}\ \ \mathsf{CH_3CH(OH)CH_3}](5164d7c9a1a3a6d4e82feb5b55249abf.png)
Гидроксимеркурирование-демеркурирование алкенов
Метод получения спиртов гидроксимеркурированием-демеркурированием алкенов имеет ряд важных преимуществ перед реакцией кислотного гидролиза[10]:
- отсутствие перегруппировок для склонных для этого субстратов;
- анти-стереоспецифичность для нормальных алкенов за исключением особых случаев пространственного затруднения;
- лучшие выходы;
- строгая ориентация по правилу Марковникова.
Механизм реакции выглядит следующим образом[11]:
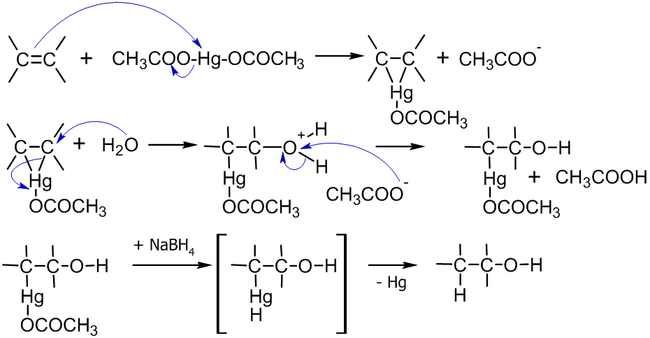
Присоединение ацетата ртути к алкену происходит по электрофильному механизму, а демеркурирование имеет радикальную природу; так как последняя стадия не обладает высокой стереоселективностью, то и весь процесс не стереоспецифичен в строгом смысле[12].
Синтез спиртов гидроксимеркурированием-демеркурированием алкенов протекает в мягких условиях, с выходами близкими к количественным (90-98 %) и практически без образования побочных продуктов; при этом промежуточное ртутьорганическое соединение не требует выделения — все стадии реакции протекают в один за другим[12].
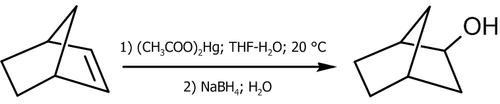
Практические примеры использования реакции (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[12]:
![\mathsf{CH_3(CH_2)_3CH\!\!=\!\!CH_2}\ \xrightarrow[2)\ NaBH_4;\ H_2O]{1)\ (CH_3COO)_2Hg,;\ THF-H_2O;\ 20^oC }\ \ \mathsf{CH_3(CH_2)_3CH(OH)CH_3}\ _{(99,5%)}](308e9f44adc430adee36cb9912d1c896.png)
![\mathsf{C_6H_5\!\!-\!\!C(CH_3)\!\!=\!\!CH_2}\ \xrightarrow[2)\ NaBH_4;\ H_2O]{1)\ (CH_3COO)_2Hg,;\ THF-H_2O;\ 20^oC }\ \ \mathsf{C_6H_5\!\!-\!\!C(CH_3)(OH)\!\!-\!\!CH_3}\ _{(100%)}](450be1b73d0d1123d9aa0c8a2881d6b2.png)
Гидроборирование алкенов с последующим окислением
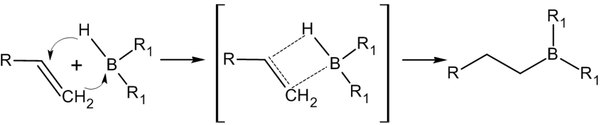
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за ее обнаружение и изучение в 1979 году ученый был удостоен Нобелевской премии по химии[13].
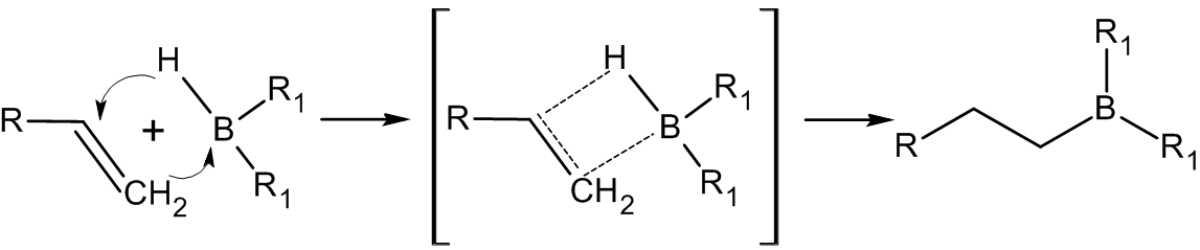
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причем присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода[14]:
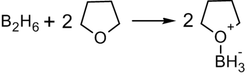
В синтезе используется, обычно, не собственно диборан, а его донорно-акцептоный комплекс с простым эфиром; а сам диборан получают реакцией борогидрида натрия с трифторидом бора в среде тетрагидрофурана[14]:

Алкилбораны легко расщепляются под действием пероксида водорода в щелочной среде, образуя спирты[14]:
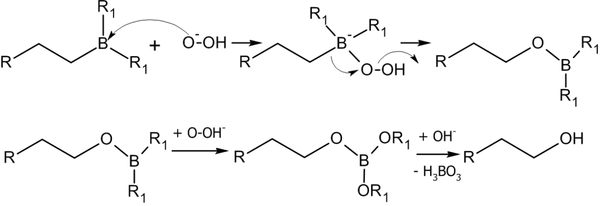
Реакция гидроборирования является реакцией син-присоединения — ее результатом становятся цис-аддукты.
Данный метод имеет широкое препаративное значение. Например, алкены с концевой двойной связью дают первичные спирты с выходами 80-90 %. Примеры практического использования метода (в скобках указаны выходы, показывающие соотношение образующихся продуктов)[7]:
![\mathsf{RCH\!\!=\!\!CH_2}\ \xrightarrow[H_2O_2,\ OH^-]{B_2H_6}\ \ \mathsf{RCH_2\!\!-\!\!CH_2OH}\ _{(94%)}+\mathsf{RCH(OH)\!\!-\!\!CH_3}\ _{(6%)}](bad9e1335d083ba48c5c812cfb67ad73.png)
![\mathsf{C_6H_5CH\!\!=\!\!CH_2}\ \xrightarrow[H_2O_2,\ OH^-]{B_2H_6}\ \ \mathsf{C_6H_5CH_2\!\!-\!\!CH_2OH}\ _{(80%)}+\mathsf{C_6H_5CH(OH)\!\!-\!\!CH_3}\ _{(20%)}](6d5405a9643b48109060da284e4ffdad.png)
![\mathsf{(CH_3)_2CHCH\!\!=\!\!CHCH_3}\ \xrightarrow[H_2O_2,\ OH^-]{B_2H_6}\ \ \mathsf{(CH_3)_2CHCH_2CH(OH)CH_3}\ _{(57%)}+\mathsf{(CH_3)_2CHCH(OH)CH_2CH_3}\ _{(43%)}](5079305bb22b1499c70a9ea3ad329a02.png)
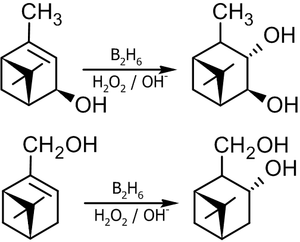
Пример синтеза бициклических терпеновых спиртов[15]:
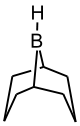
Для повышения селективности реакции гидроборирования используют замещенные, пространственно затрудненные бораны[16]:
Тексилборан
2,3-диметилбутил-2-боранДисиамилборан
бис-(1,2-диметилпропил)боран9-ББН
9-борабицикло[3,3,1]нонанДиизопинокамфеилборан
(+)3-дипинанилборан


Использование, например, в реакции со стиролом дисиамилборана (DIAB) повышает выход первичного спирта с 80 % до 98 %[17]:
![\mathsf{C_6H_5CH\!\!=\!\!CH_2}\ \xrightarrow[H_2O_2,\ OH^-]{DIAB}\ \ \mathsf{C_6H_5CH_2\!\!-\!\!CH_2OH}\ _{(98%)}+\mathsf{C_6H_5CH(OH)\!\!-\!\!CH_3}\ _{(2%)}](5e815b8bf48fe8fd320929ee4f077bbd.png)
Высокая селективность указанных выше производных борана позволяет избирательно вступать в реакцию с цис-изомером, находящемся в смеси транс-изомером или гидроборировать одну двойную связь из двух имеющихся в молекуле алкена, например[6]:
![\mathsf{CH_2\!\!=\!\!CH\!\!-\!\!(CH_2)_3\!\!-\!\!C(CH_3)\!\!=\!\!CH_2}\ \xrightarrow[H_2O_2,\ OH^-]{(Sia)_2BH}\ \ \mathsf{HOCH_2\!\!-\!\!CH_2\!\!-\!\!(CH_2)_3\!\!-\!\!C(CH_3)\!\!=\!\!CH_2}](530cf323c8557b30df8b8365d67fbb64.png)
Гидроборирование алкенов с последующим присоединением окиси углерода
Одним из лучших способов получения третичных спиртов является присоединение к окиси углерода к алкилборанам. Реакция легко протекает при обычном давлении и температуре около 125 °C, в качестве растворителя используется диглим[7]:

![\mathsf{(RCH_2\!\!-\!\!CH_2-)_3B+CO}\xrightarrow{125^oC}\mathsf{[(RCH_2\!\!-\!\!CH_2-)_3B^-\!\!\!\!-\!\!C}\!\!\equiv\!\!\mathsf{O^+]}\rightarrow\mathsf{[(RCH_2\!\!-\!\!CH_2-)_2B\!\!-\!\!CO\!\!-\!\!CH_2CH_2R]}\rightarrow](8594bae938563a067a37c73dfc011fda.png)
![\rightarrow\mathsf{[(RCH_2\!\!-\!\!CH_2-)_3C\!\!-\!\!B\!\!=\!\!O]}\xrightarrow[NaOH,\ H_2O]{H_2O_2}\mathsf{(RCH_2\!\!-\!\!CH_2-)_3COH+Na[B(OH)_4]}](b229b97e611b465c61ac9d3ccd17d25c.png)
Этот метод дает неплохие выходы со многими алкенами[14]:
![\mathsf{CH_3CH\!\!=\!\!CHCH_3}\xrightarrow[3)\ H_2O_2,\ NaOH,\ H_2O]{1)\ B_2H_6;\ 2)\ CO}\mathsf{(CH_3CH_2CH(CH_3)-)_3COH}\ _{(87%)}](8ee4638429d0d3fdaa01f6e04e4d6a9b.png)
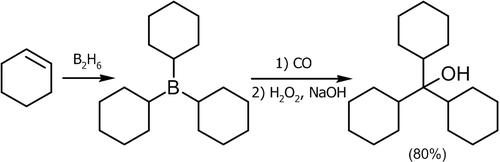
Если данную реакцию проводить в присутствии водного раствора щелочи, получаются вторичные спирты[18]:

Гидроформилирование алкенов
Широко используемая в промышленности классическая реакция гидроформилирования алкенов, то есть каталитического присоединение к ним водорода и монооксида углерода с получением на выходе альдегидов[19], может быть проведена таким образом, что продуктом её реакции сразу будут спирты без выделения промежуточных карбонильных соединений Это метод иногда называют восстановительным гидроформилированием.

Катализирует реакцию координационно ненасыщенный гидрокарбонил кобальта, образующийся в ходе реакции[19]:


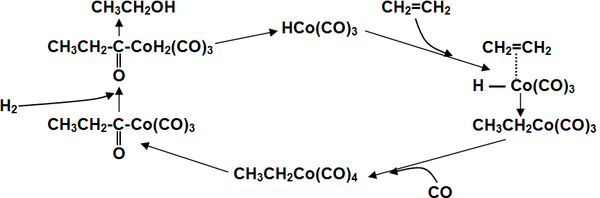
Для получения спиртов в одну стадию в качестве катализаторов используют карбонилы кобальта, модифицированные фосфинами, что помимо более активного гидрирования, позволяет добиться существенно более высокой селективности выхода нормальных продуктов (до 90 %) вследствие стерического эффекта объемного фосфинового лиганда в переходном состоянии[20].
Получение спиртов из простых эфиров и спиртов
Реакция гомологизации спиртов
Гомологизация, то есть превращение органического соединения в свой гомолог путём внедрения одной или нескольких метиленовых групп, для спиртов была впервые осуществлена в 1940 году — на основе метанола каталитическим путём под воздействием высокого давления был синтезирован этанол[21]:

Реакция гомологизации по своему механизму близка реакции гидроформилирования алкенов и в настоящее время с помощью модифицированных катализаторов кобальта и рутения и добавления йодид-ионов в качестве промоторов удаётся добиться 90 % выхода по этанолу[21].
Исходный метанол также получают из окиси углерода (катализаторы на основе оксидов меди и цинка, давление 5-10 МПа, температура 250 °C)[21], так что общая схема выглядит следующим образом:
![\mathsf{C+H_2O}\rightarrow\mathsf{CO+H_2}\rightarrow\mathsf{CH_3OH}\ \xrightarrow[-\ H_2O]{CO\ +\ H_2}\ \ \mathsf{CH_3CH_2OH}](36f036763974bdee75aa6d43065ceb2b.png)
Побочными продуктами реакции в случае синтеза этанола будут ацетальдегид, этилен и диэтиловый эфир.
Реакция Гербе́
Реакция Гербе представляет собой высокотемпературный (200 °C, давление 5—6 МПа) процесс каталитической конденсации первичных алифатических спиртов, не имеющих разветвления в α-положении, по следующей схеме[22]:

В качестве катализаторов используют сложную смесь на основе никеля Ренея, меди, солей железа и других компонентов[23].
Предполагаемый условный механизм реакции[23]:



или

Реакция имеет ограниченное применение как из-за жестких условий её проведения и относительно низкого выхода (как правило, до 70 %), так и образования кислоты и альдегида в качестве побочного продукта[23].
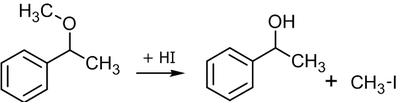
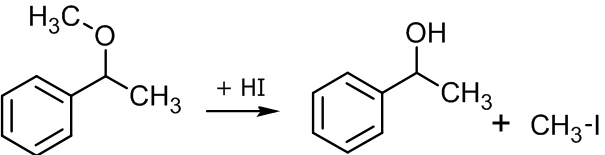
Кислотное расщепление простых эфиров
В лабораторной практике подобный способ получения спиртов крайне редок, так как именно спирты служат исходным компонентом для синтеза простых эфиров. Вместе с тем, если в качестве исходного объекта выбран, скажем природный простой эфир сложной структуры, его лабораторное расщепление до исходного спирта может оказаться востребованным. Кроме того, в некоторых случаях для защиты гидроксильной группы в процессе многоступенчатого синтеза, её могут перевести в эфирную и вводить в реакцию уже простой эфир. По окончании процесса для обратного превращения соединения в спирт может потребоваться расщепление эфира (см. подробнее подраздел «Защита через простые эфиры»).
Обычно реакцию проводят нагреванием эфира и концентрированного раствора бромоводородной или йодоводородной кислоты, при этом расщепление может осуществляться как по механизму SN1, так и SN2[24]:
 SN1 механизм расщепления простых эфиров
SN1 механизм расщепления простых эфиров
 SN2 механизм расщепления простых эфиров
SN2 механизм расщепления простых эфировЕсли в реакцию вступают несиметричные эфиры, в результате получаются два спирта и два галогенпроизводных, однако если эфир метиловый, продуктом реакции будет спирт и метилиодид или метилбромид[25]:
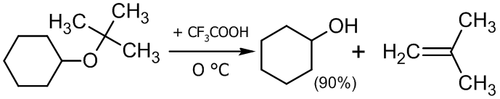
Для расщепления эфиров могут использоваться также кислоты Льюиса: BF3, BCl3, AlCl3 и др[25], а также сильные органические кислоты. Например, расщепление трет-бутилциклогексилового эфира трифторуксуной кислотой происходит по механизму SN1 с образованием циклогексанола и 2-метилпропена[26]:
Перегруппировка Виттига
Простые эфиры под действием фениллития перегруппировываются в спирты (Георг Виттиг, 1942 год):
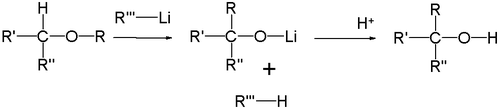
Реакция представляет собой карбанионную перегруппировку, которая осуществляется через радикальный механизм расщепления-рекомбинации[27]:
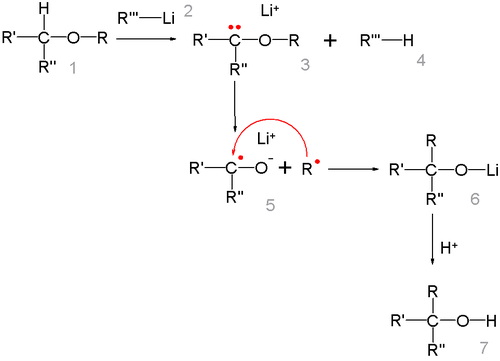
Говоря о стереохимии перегруппировки Виттига, следует отметить, что образование новой C-C связи происходит настолько быстро, что радикал R не успевает инвертироваться, поэтому обычно, реакция протекает с сохранением исходной конфигурации[28]. Изучение перегруппировки на примере β-алкоксиалкилаллиловых эфиров (общий вид:
 ) показало, что в результате реакции с выходом 14-32 % образовывались син-1,3-диол производные с селективностью 90-95 %[29].
) показало, что в результате реакции с выходом 14-32 % образовывались син-1,3-диол производные с селективностью 90-95 %[29].Перегруппировка Виттига может осуществляться не только с помощью алкил- или ариллитиевых соединений (фениллитий, бутиллитий, метиллитий, диэтиламид лития и пр.), но и под действием других сильных оснований; например, следующая реакция протекает в жидком аммиаке в присутствии амида калия (выход 90 %)[30]:
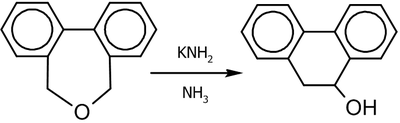
Для аллильно-замещенных субстратов (1,2)-перегруппировка конкурирует с (2,3)-перегруппировкой, которую можно наблюдать практически полностью независимо при низких температурах[27]:
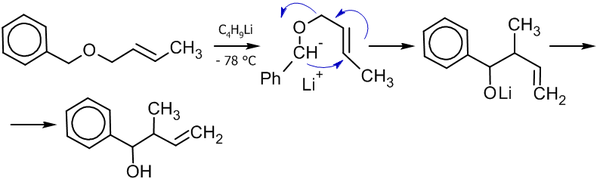
Получение спиртов из альдегидов и кетонов
В данном разделе помимо получения собственно спиртов из альдегидов и кетонов приведены синтезы гидроксикарбонильных соединений (кетоспирты и производные гидроксикарбоновых кислот; смотри подразделы «Альдольная конденсация», «Бензоиновая конденсация», «Реакция Реформатского», «Реакция Иванова»). Это связано с тем, что приведенные реакции являются мощными препаративными методами и широко используются на практике.
Этинилирование карбонильных соединений
Важным методом получения ацетиленовых спиртов является реакция Фаворского, иначе говоря, реакция этинилирования карбонильных соединений:


В реакцию вступают незамещенные алкины (берутся в большом избытке), кетоны и некоторые альдегиды (чаще всего используется формальдегид) в присутствии оснований (KOH или NaNH2 в органическом растворителе) при температуре от −70 до +40 °C, давлении 0,4-0,9 МПа[31].
Механизм данной реакции связан с нуклеофильным присоединеним этинильного карбаниона к карбонильной группе[32]:
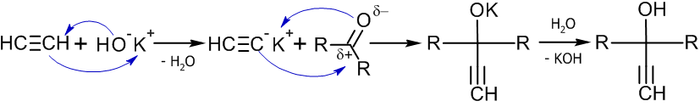
Примеры реакций[33]:


Существуют, по-меньшей мере, две модификации этого метода:
- Реакция Реппе — конденсация алкинов с альдегидами или кетонами в присутствии каталитических количеств ацетиленидов меди, серебра или ртути[34].
- Реакция Нефа — реакция с последующим гидролизом ацетиленидов щелочных металлов с кетонами, включая α,β-непредельные и ароматические карбонильные соединения[35].
Взаимодействие альдегидов с аллилборанами
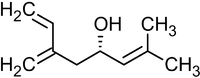
Современным методом получения аллильных спиртов заданной конфигурации является применение в качестве агента аллилборана, который реагирует с альдегидами в присутствии оснований по следующей схеме[36]:

Реакция была использована, в частности, в качестве базовой для полного синтеза ряда природных соединений и их аналогов, например, феромонов короеда[36]:
Ипсенол Ипсдиенол 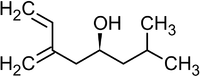
Существуют различные методики этой реакции, среди которых:
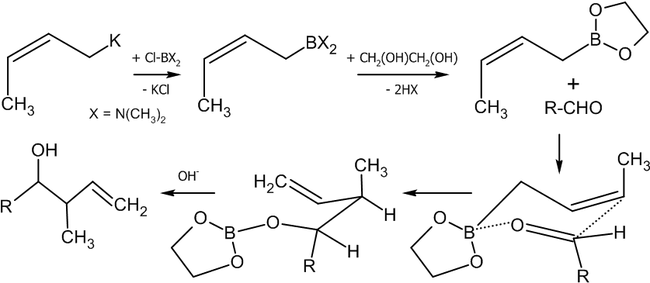
Если в реакцию вступают цис-алкены, в основном, будут образовываться син-продукты присоединения (97 % от общего выхода).
Другим удобным агентом, реагирующим по данной схеме является аллилборный эфир винной кислоты (Рауш, 1985 год)[38].
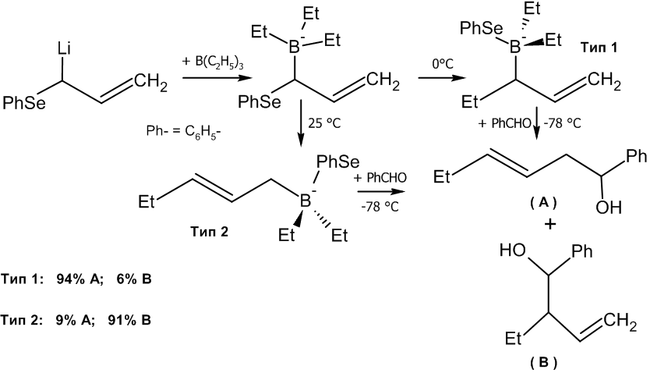
Как видно из схемы, меняя температуру реакции, можно вызвать миграцию атома бора к соседнему углероду, получив тот или иной изомерный спирт.
Реакция Сакураи
Другой способ аллилирования, заключающийся в электрофильном взаимодействии аллилсиланов с различными соединениями в присутствии кислот Льюиса носит название реакции Сакураи. С точки зрения получения спиртов, существуют две модификации подобного синтеза[39]:
- Реакция с карбонильными соединениями с получением вторичных или третичных спиртов:
![\mathsf{RCOR'+CH_2\!\!=\!\!CHCH_2Si(CH_3)_3}\ \xrightarrow[H_2O]{Lewis\ acid}\ \mathsf{RR'C(OH)CH_2CH\!\!=\!\!CH_2}](3cad4ae85fa877af1d8f935ce93cc870.png)
- Реакция с эпоксидами с получением вторичных спиртов:
![\mathsf{(CHR\!\!-\!\!CHR)O+CH_2\!\!=\!\!CHCH_2Si(CH_3)_3}\ \xrightarrow[H_2O]{Lewis\ acid}\ \mathsf{RCH(OH)CH(R)CH_2CH\!\!=\!\!CH_2}](4573b44f613144e6481d4dbd0c07a20d.png)
В качестве катализаторов реакции могут выступать: TiCl4, BF3, SnCl4 и пр.
Пример практического использования реакции Сакураи[40]:
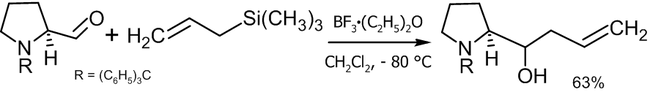
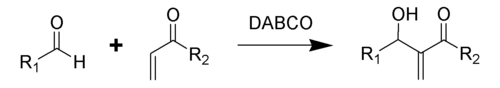
Реакция Бэйлиса-Хиллмана-Морита
Традиционная реакция Бэйлиса-Хиллмана-Морита представляет собой метод получения аллиловых кетоспиртов взаимодействием альдегидов с метилвинилкетонами или другими активированными алкенами в присутствии третичных фосфинов и каталитических количеств фенола или его производных[41]. Впоследствии реакция была несколько модифицирована: в качестве катализатора стали использовать третичные амины (например: 1,4-диазобицикло[2.2.2]октан или DABCO[42]):
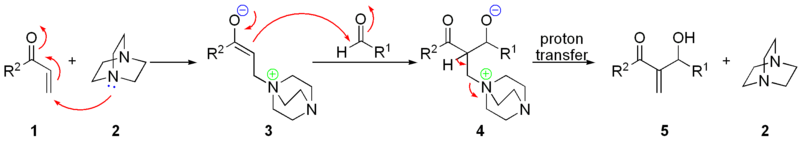
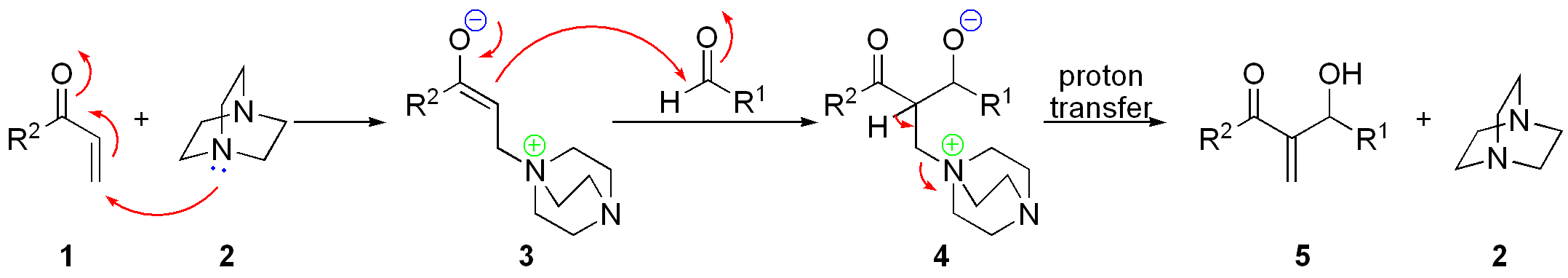
Предположительно, механизм реакции выглядит следующим образом[43]:
Реакция Нозаки-Хияма-Киши
Реакция Нозаки-Хияма-Киши представляет собой современный метод получения спиртов селективным восстановительным сочетанием альдегидов с винил- или аллилгалогенидами (бромиды или йодиды) в присутствии хром-никелевого катализатора[44]:
![\mathsf{R'\!\!-\!\!CHO+R\!\!-\!\!CH\!\!=\!\!CHBr}\ \xrightarrow[(CH_3)_2S=O]{CrCl_2,\ NiBr_2}\ \mathsf{R\!\!-\!\!CH\!\!=\!\!CH\!\!-\!\!CH(OH)R'}](6e3ecdb0758fcccd87b0d6d178132aa9.png)
Каталитический цикл реакции выглядит следующим образом[45]:
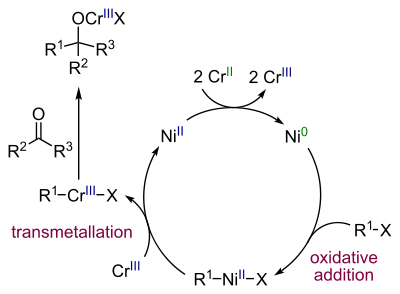
Каталитическое сочетание альдегидов с аллиловыми спиртами и их производными
Аналогом синтетического метода, рассмотренного в предыдущем подразделе, является реакция сочетания альдегидов с аллиловыми спиртами и их производными в присутствии катализаторов. В научной литературе описано множество лабораторных методик осуществления подробного синтеза с примененением органических соединений кремния, олова, хрома, лития, рутения, палладия, цинка, титана, циркония и других металлов.
Приведём некоторые характерные примеры использования этого метода на практике:
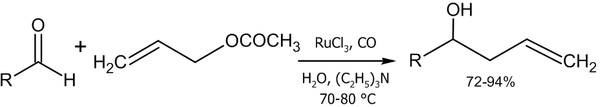
- Реакция аллилацетата с альдегидами в присутствии солей рутения (Denmark S. E., Nguyen S. T., 2009 год)[46]:
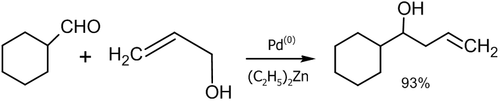
- Реакция аллилового спирта с алифатическими альдегидами в присутствии палладиевого катализатора (Masanari Kimura, Masamichi Shimizu, Kazufumi Shibata, Minoru Tazoe, Yoshinao Tamaru, 2003 год)[47]:
- Реакция аллилтрибутилстанната с альдегидами в присутствии рениевого комплекса (Yutaka Nishiyama, Fujio Kakushoua, Noboru Sonoda, 2004 год)[48]:
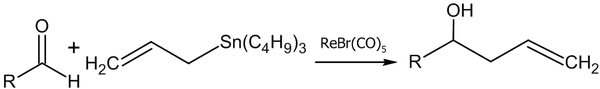
Более подробно о современных методах получения спиртов реакцией аллиловых спиртов и их производных с карбонильными соединениями можно прочитать в монографии: Junzo Otera. Modern Carbonyl Chemistry — Wiley-VCH, Weinheim, 2000—613 Pages — ISBN 978-3-527-29871-6.
Реакция Канниццаро
Реакция Канниццаро представляет собой оксилительно-восстановительное диспропорционирование альдегидов в первичные спирты и карбоновые кислоты под действием оснований[49]:

На первом этапе реакции происходит нуклеофильное присоединение основания (например: гидроксид-аниона) к карбонильному углероду альдегида. Образующийся анион депротонируется (это требует воздействия достаточно сильного основания) с образованием промежуточного дианиона, который затем вступает в реакцию с молекулой альдегида:
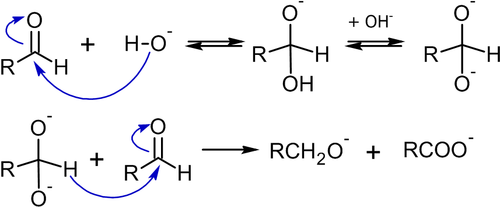
В реакцию Канниццаро вступают альдегиды, не способные к енолизации (не имеющие α-водорода), поскольку для последних преобладающей будет альдольная конденсация. Например, в известном примере с бензальдегидом, выход бензилового спирта может достигать 90 %[50]:

Чаще реакция Канниццаро используется для синтеза ароматических и гетероароматических спиртов[50].
Для повышения реакционного диапазона используемых альдегидов и увеличения выхода спиртов на практике используется перекрестная реакция Канниццаро, то есть использование двух различных альдегидов, причем в качестве альдегида-восстановителя обычно используется формальдегид, который в ходе реакции окисляется в муравьиную кислоту[51]:

В настоящее время существуют более эффективные синтетические методы, поэтому полезность реакции Канниццаро ограничена, как правило, диспропорционированием кетоальдегидов в гидроксикарбоновые кислоты[52]:

Циангидринный синтез
Карбонильные соединения, особенно альдегиды и стерически не затруднённые кетоны, легко вступают в реакции нуклеофильного присоединения c цианистым водородом (нуклеофил CN−) с образованием циангидринов[53]:

Для ароматических кетонов вместо HCN используют цианид диэтилалюминия (C2H5)2AlCN или цианотриметилсилан (CH3)3SiCN, продукт присоединения которого затем гидролизуется до циангидрина[53]:

Далее при необходимости циангидрин легко гидролизуется до гидроксикислоты или восстанавливается в аминоспирт:


Альдольная конденсация
Альдольная конденсация — это одна из старейших реакций органического синтеза (1872 год, Вюрц), в которой две молекулы альдегида или кетона под действием основания или кислоты соединяясь, образуют кетоспирты или альдоли[54]:
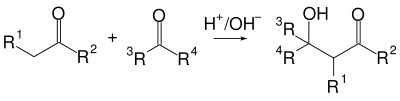
Возможны два механизма этой реакции: щелочной или кислотный, однако с точки зрения синтеза спиртов, последний менее предпочтителен, так как часто реакция не останавливается на стадии спирта а протекает дальше с дегидратацией и образованием непредельных карбонильных соединений (кротоновая конденсация)[54].
Механизм конденсации, происходящий под действием основания, следующий[54]:
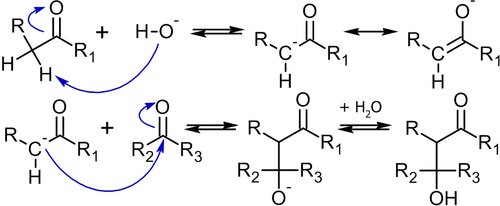
Для проведения конденсации, как видно из её механизма, необходимо, чтобы хотя бы одна из молекул содержала водород в α-положении к карбонильной группе. Обычно, для отрыва этого атома водорода силы гидроксид-иона бывает достаточно, но в отдельных случаях используют и более сильные основания, например — бутиллитий.
Возможны пять комбинаций протекания альдольной конденсации[53]:
- Взаимодействие двух молекул одного альдегида: реакция легко осуществима и приводит к одному продукту, однако из-за электроноакцепторного эффекта альдегидной группы, использование их для синтеза спиртов не эффективно из-за обычно преобладающего расщепления образующегося альдоля до непредельного альдегида, например:

- Взаимодействие двух молекул различных альдегидов: теоретически реакция может привести к четырем разным продуктам, но если один из альдегидов не будет содержать α-карбонильный водород, возможно осуществление только перекрестной реакции;
- Взаимодействие двух молекул одного кетона: реакция сильно смещена влево, поэтому для её проведения либо пользуются специальным оборудованием (аппарат Сокслета), позволяющим фактически удалять из реакционной зоны продукт реакции или использовать в качестве основания особые реагенты (например: нитрид бария);
- Взаимодействие двух молекул разных кетонов: применяется довольно редко и в случаях, когда один из кетонов не содержит α-карбонильный водород;
- Взаимодействие одной молекулы альдегида с одной молекулой кетона: чаще всего в качестве альдегида используют формальдегид, который даёт один продукт конденсации. Другой вариант — использование не самого кетона, а его енольной формы, например в виде литиевой соли или силилового эфира.
Кроме собственно альдегида, возможно использование имина и диизопропиламида лития в качестве основания[53]:
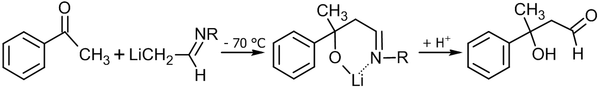
Разновидностью альдольной реакции является реакция Мукаямы, в которой используются силиленольные эфиры:
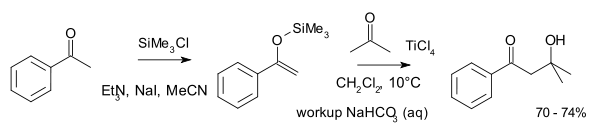
Бензоиновая конденсация
Бензоиновая конденсация представляет собой обратимое образование из альдегидов (преимущественно — ароматических) под действием цианид-ионов CN− α-оксикетонов (ацилоинов) с общей формулой —СR(ОН)—С(O)О—:

В этой реакции на первом этапе цианид анион вступает в реакцию нуклеофильного присоединения с альдегидом. Перегруппировка образующегося интермедиата в карбанион завершается дальнейшим присоединением второй карбонильной группы также по нуклеофильному механизму. Завершает реакцию переноса протона и отщепление цианидной группы с образованием бензоина в качестве конечного продукта.
Реакция Иванова
Реакция Иванова представляет собой метод получения β-гидроксикарбоновых кислот общей формулой —CR(OH)—CR1(COOH)— из карбонильных соединений и реактивов Иванова: магнийгалогенпроизводных солей арилуксусных кислот (обычно — фенилуксусной кислоты)[55]. Например:
![\mathsf{R\!\!-\!\!CO\!\!-\!\!R+[Ar\!\!-\!\!CH^-\!\!\!\!-\!\!COONa]MgCl^+}\rightarrow\mathsf{[Ar\!\!-\!\!CR(COONa)\!\!-\!\!CHR(OMgCl)]}\rightarrow\mathsf{Ar\!\!-\!\!CR(COOH)\!\!-\!\!CHROH}](aa1cf2007263e5eb586b7b485b6de9c0.png)
При необходимости оксикислота может быть легко превращена в соответствующий спирт методом декарбоксилирования.
Получение спиртов из карбоновых кислот и сложных эфиров
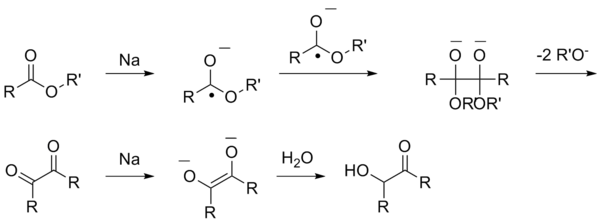
Гидролиз сложных эфиров карбоновых кислот

Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Ацилоиновая конденсация
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Декарбонилирование карбоновых кислот
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение спиртов восстановлением эпоксидов и карбонильных соединений
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление эпоксидов гидридами металлов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление карбонильных соединений гидридами металлов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление карбонильных соединений по реакции Меервейна-Пондорфа-Верлея
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление карбонильных соединений органическими реагентами
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление ароматических кетонов щелочными металлами
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление карбоновых кислот и сложных эфиров по методу Буво-Блана
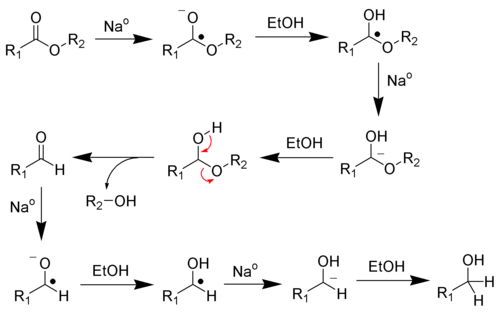
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Восстановление хлорангидридов карбоновых кислот
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Каталитическое гидрирование карбонильных соединений
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение спиртов с использованием металлорганических соединений
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Присоединение реактивов Гриньяра к эпоксидам
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Присоединение реактивов Гриньяра к альдегидам или кетонам
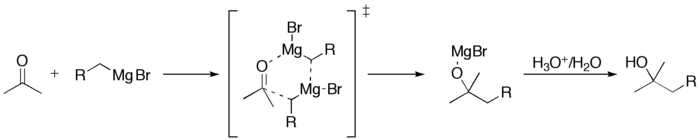
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Присоединение реактивов Гриньяра к сложным эфирам или ацилгалогенидам
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение спиртов окислительными методами
Окисление алканов и циклоалканов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Окисление алкенов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Озонолиз алкенов с последующим восстановлением
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Реакция Циглера
Является методом синтеза высших спиртов (С8 и выше) при помощи алюминийорганических соединений. Алюминийорганические соединения легко могут быть получены из олефинов в присутствии водорода. Данным методом можно получать чистые первичные спирты
![\mathsf{RCH\!\!=\!\!CH_2}\ \xrightarrow[-RH]{H_2,\ AlR_3}\ \mathsf{RCH_2CH_2\!\!-\!\!AlR_2}\ \xrightarrow{O_2}\ \mathsf{RCH_2CH_2\!\!-\!\!O\!\!-\!\!Al(OR)_2}\ \xrightarrow{H_2O}\ \mathsf{RCH_2CH_2OH}](50f0829c96b4715f2d59ff76bf4d048c.png)
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Окисление реактивов Гриньяра
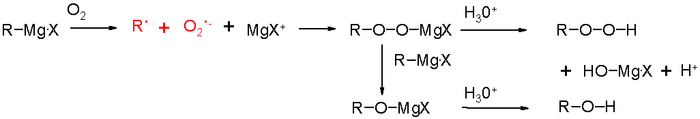
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Окисление Тамао-Кумада-Флеминга
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Прочие методы получения спиртов
Гидролиз эфиров неорганических кислот
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Диазотирование первичных алифатических аминов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Реакция Демьянова
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Реакция Фриделя-Крафтса
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.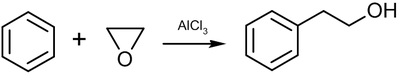
Восстановление сложных эфиров тиокислот
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Реакция Кулинковича
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Краткий обзор методов органического синтеза многоатомных спиртов
Получение 1,2-диолов гидратацией эпоксидов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение 1,2-диолов конденсацией карбонильных соединений
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение 1,2-диолов цис-дигидроксилированием алкенов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение 1,2-диолов транс-дигидроксилированием алкенов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение 1,3-диолов по реакции Принса
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение многоатомных спиртов по реакции Толленса
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Получение спиртов в промышленности
Получение спиртов методами основного органического синтеза
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленные синтезы на основе окиси углерода
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленное получение спиртов гидратацией алкенов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленное получение спиртов окислением углеводородов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленное получение спиртов гидрированием карбонильных соединений
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленное получение спиртов щелочным гидролизом галогенуглеводородов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Химические и биохимические методы получения спиртов из природного сырья
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленное получение спиртов щелочным гидролизом природного сырья
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Биохимическое гидролизное получение спиртов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Промышленный биосинтез спиртов
Этот раздел не завершён. Вы поможете проекту, исправив и дополнив его.Примечания
- ↑ Спирты // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 800.
- ↑ 1 2 Курц А.Л., Брусова Г.П., Демьянович В.М. Получение одноатомных спиртов. Одно- и двухатомные спирты, простые эфиры и их сернистые аналоги. ChemNet. Химический факультет МГУ (1999). Архивировано из первоисточника 23 апреля 2012. Проверено 1 сентября 2009.
- ↑ 1 2 3 4 5 Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 586-623 с. — ISBN 5-94628-171-2
- ↑ Глава 3.3.3. Нуклеофильное замещение галогена // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 660-661.
- ↑ 1 2 Глава 4.1.1. Одноатомные спирты // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 13-118.
- ↑ 1 2 3 Mарч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 132-212.
- ↑ 1 2 3 Бюлер К., Пирсон Д. Глава 4. Спирты. Б. Реакции присоединения и замещения // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 213-219.
- ↑ 1 2 Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидратация алкенов. Алкены (Часть 1). ChemNet. Химический факультет МГУ (1998). Архивировано из первоисточника 23 апреля 2012. Проверено 2 сентября 2009.
- ↑ Лебедев Химия и технология основного органического и нефтехимического синтеза: Учебник для вузов / Пер. с англ. — 4-е изд. перераб. и доп.. — М.: «Химия», 1988. — С. 180-184. — ISBN 5-7245-0008-6
- ↑ Глава 2.2.3. Реакции олефинов // Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 207-208.
- ↑ Carey F.A. Oxymercuration-Demercuration of Alkenes (англ.). Organic chemistry. McGraw-Hill Higher Education. Архивировано из первоисточника 23 апреля 2012. Проверено 2 сентября 2009.
- ↑ 1 2 3 Реутов О.А, Курц А.Л., Бутин К.П. Органическая химия. — М.: Издательство МГУ, 1999. — Т. 1. — С. 385-387. — ISBN 5-211-03054-0
- ↑ The Nobel Prize in Chemistry 1979 (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. Архивировано из первоисточника 22 августа 2011. Проверено 3 сентября 2009.
- ↑ 1 2 3 4 Курц А Л., Ливанцов М.В., Ливанцова Л.И. Гидроборирование алкенов. Алкены (Часть 2). ChemNet. Химический факультет МГУ (1998). Архивировано из первоисточника 23 апреля 2012. Проверено 2 сентября 2009.
- ↑ Кучин А.В., Фролова Л.Л., Пантелеева М.В. Бициклические терпеновые диолы как лиганды для синтеза хиральных катализаторов (pdf). Англо-русскоязычный общественный химический журнал «Бутлеровские сообщения». Архивировано из первоисточника 23 апреля 2012. Проверено 31 августа 2009.
- ↑ Борорганические соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 594-603.
- ↑ Вацуро К.В., Мищенко Г.Л. 109. Браун (Brown H.C.) // Именные реакции в органичнеской химии. — М.: «Химия», 1976. — С. 76-77.
- ↑ Дядченко В.П., Трушков И.В., Брусова Г.П. Синтетические методы органической химии. Части 1-2. — М.: МГУ им. М.В. Ломоносова, 2004. — С. 46.
- ↑ 1 2 Караханов Э.А. Синтез-газ как альтернатива нефти. I. Процесс Фишера-Тропша и оксо-синтез // Соросовский образовательный журнал. — 1997. — № 3. — С. 73-74.
- ↑ Шелдон Р.А. Химические продукты на основе синтез-газа = Chemicals From Synthesis Gas / Под ред. С.М.Локтева. — М.: «Химия», 1987. — С. 92.
- ↑ 1 2 3 Караханов Э.А. Синтез-газ как альтернатива нефти. II. Метанол и синтезы на его основе // Соросовский образовательный журнал. — 1997. — № 12. — С. 68.
- ↑ Гербе реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 1024-1025.
- ↑ 1 2 3 Бюлер К., Пирсон Д. Глава 4. Спирты. Ж. Присоединение карбанионов // Органические синтезы = Survey of Organic Syntheses / Пер. с англ. — М.: «Мир», 1973. — Т. 1. — С. 268-279.
- ↑ Травень В.Ф. Глава 18. Простые эфиры. Циклические эфиры // Органическая химия: Учебник для вузов: В 2 т. / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 2. — 97-98 с. — ISBN 5-94628-172-0
- ↑ 1 2 Mарч Дж. Глава 10. Реакции алифатического нуклеофильного замещения // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 2. — С. 11-240.
- ↑ McMurry J. Organic chemistry. — Seven edition. — Thomson, 2008. — P. 604, 658. — ISBN 0-495-11258-5
- ↑ 1 2 (1,2)-Wittig Rearrangement (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 23 апреля 2012. Проверено 14 сентября 2009.
- ↑ Бутин К.П. Механизмы органических реакций: достижения и перспективы // Журнал Российского химического общества им. Д.И.Менделеева. — 2001. — Т. XLV. — № 2. — С. 32.
- ↑ Schreiber S.L., Goulet M. T. Stereochemistry of the 1,2-Wittig Rearangement: A Synthesis of syn-1,3-diol Monoethers (англ.) // Tetrahedron Letters. — 1987. — Т. 28. — № 10. — С. 1043-1046.
- ↑ Физер Л., Физер М. Реагенты для органического синтеза = Reagents For Organic Synthesis / Под редакцией проф. И.Л.Кнунянца и д.х.н. Р.Г.Костяновского. — М.: «Мир», 1970. — Т. 1. — С. 56-57.
- ↑ Фаворского реакции // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 95-96.
- ↑ Темкин О. Н. Химия ацетилена. «Ацетиленовое дерево» в органической химии XXI века // Соросовский образовательный журнал. — 2001. — Т. 7. — № 6. — С. 39.
- ↑ Вацуро К.В., Мищенко Г.Л. 595. Фаворский // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 411-412.
- ↑ Вацуро К.В., Мищенко Г.Л. 506. Реппе (Reppe) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 351.
- ↑ Вацуро К.В., Мищенко Г.Л. 424. Неф (Nef) // Именные реакции в органической химии. — М.: «Химия», 1976. — С. 296.
- ↑ 1 2 Бубнов Ю.Н. Аллилбораны. Принципы реагирования и применение в органическом синтезе // Вестник Московского университета : Серия 2. Химия. — 2005. — Т. 46. — № 3. — С. 140-144.
- ↑ 1 2 Reich I.L. Allylborane Reactions (англ.). Chemistry 842 - Fall 2004 Course Outline. University of Wisconsin. Department of Chemistry. Проверено 15 сентября 2009.
- ↑ Ли Дж. Роуш (Roush). Аллилборонат как реагент // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 306. — ISBN 5-94774-368-X
- ↑ Hosomi-Sakurai Reaction (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 23 апреля 2012. Проверено 20 сентября 2009.
- ↑ Li J. J., Limberakis C., Pflum D. A. Modern organic synthesis in the laboratory: a collection of standard experimental procedures. — New York: Oxford University Press, Inc, 2007. — P. 139. — ISBN 978-0-19-518798-4
- ↑ Traditional Morita-Baylis-Hillman reaction of aldehydes with methyl vinyl ketone co-catalyzed by triphenylphosphine and nitrophenol (англ.). Abstracts. Organic Chemistry Portal. Архивировано из первоисточника 23 апреля 2012. Проверено 23 сентября 2009.
- ↑ Baylis-Hillman Reaction (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 21 августа 2011. Проверено 23 сентября 2009.
- ↑ Smith A.C. Morita Baylis Hillman Reaction (англ.) (pdf). New Methodology and Synthesis of Natural Product. The University of North Carolina at Chapel Hill. Архивировано из первоисточника 23 апреля 2012. Проверено 23 сентября 2009.
- ↑ Ли Дж. Нозаки-Хияма-Киши (Nozaki-Hiyama-Kishi). Реакция // Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — С. 249. — ISBN 5-94774-368-X
- ↑ Kallemeyn J. M. The Nozaki-Hiyama-Kishi reaction (англ.) (pdf). The Department of Chemistry at the University of Illinois at Urbana-Champaign (12 April 2002). Архивировано из первоисточника 23 апреля 2012. Проверено 15 сентября 2009.
- ↑ Catalytic, Nucleophilic Allylation of Aldehydes with Allyl Acetate (англ.). Organic Letters. ACS Publications. Архивировано из первоисточника 23 апреля 2012. Проверено 16 сентября 2009.
- ↑ Pd-Catalyzed Nucleophilic Alkylation of Aliphatic Aldehydes with Allyl Alcohols: Allyl, 2-Tetrahydrofuryl, and 2-Tetrahydropyranyl Ethers as Useful C3, C4, and C5 Sources (англ.). Angewandte Chemie. Wiley InterScience. Архивировано из первоисточника 23 апреля 2012. Проверено 16 сентября 2009.
- ↑ Nishiyama Y., Kakushou F., Sonoda N. Rhenium complex-catalyzed allylation of aldehydes with allyltributylstannane (англ.) // Tetrahedron Letters. — 2005. — Т. 46. — № 5. — С. 787-789.
- ↑ Канниццаро реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 603-604.
- ↑ 1 2 Глава 5.3.7. Реакция Канниццаро // Общая органическая химия. Кислородсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1982. — Т. 2. — С. 737-739.
- ↑ Mарч Дж. Глава 19. Реакции окисления и восстановления // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 4. — С. 337-338.
- ↑ Cannizzaro Reaction (англ.). Name Reactions. Organic Chemistry Portal. Архивировано из первоисточника 23 апреля 2012. Проверено 23 сентября 2009.
- ↑ 1 2 3 4 Mарч Дж. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: «Мир», 1988. — Т. 3. — С. 324-427.
- ↑ 1 2 3 Альдольная конденсация // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 202-204.
- ↑ Иванова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 344—345.
Категория:- Спирты


























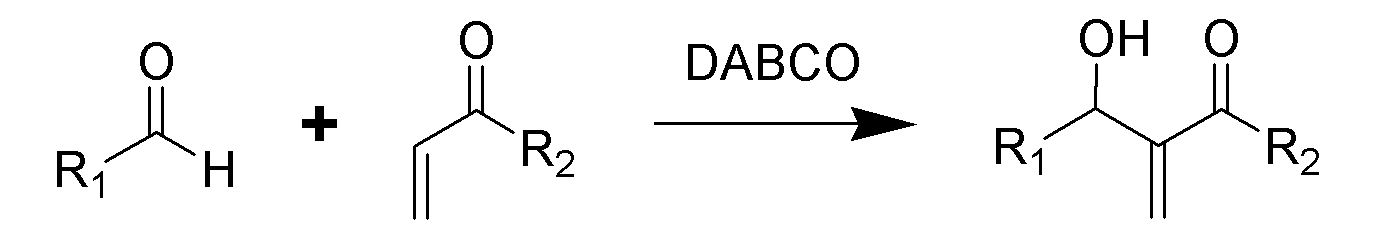















Wikimedia Foundation. 2010.