- АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
А. МОНОФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
1. С1-: металлоорганические соединения. Эти соединения обычно получают двумя методами: а) действием активного металла (Na, Li, Mg, Zn) на органический галогенид, например:

или б) действием галогенида менее активного металла на металлоорганическое соединение более активного металла, например:
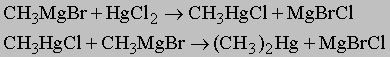
Металлоорганические соединения обычно называют, ставя на первое место название радикала и прибавляя к нему название металла (или соли металла), к которому присоединен радикал, например, метилнатрий CH3Na; диэтилцинк (C2H5)2Zn; этилртутьхлорид C2H5HgCl. Вообще говоря, реакционная способность этих соединений возрастает с ростом активности металла; так, реакции алкилпроизводных цинка или ртути протекают медленнее, чем реакции алкилпроизводных магния или натрия. Алкилпроизводные щелочных металлов (Li, Na, K) можно приготовить взаимодействием свободного металла с алкилгалогенидами или с диалкилртутью:

Метод (а) (см. выше) можно использовать для натрия и калия только в таком инертном растворителе, как пентан. Тонко раздробленный металл должен присутствовать в большом избытке, а реакционную смесь необходимо очень сильно перемешивать, в противном случае реакция

разрушает металлалкил по мере его образования. Для синтетической органической химии очень ценны алкилмагнийгалогениды RMgX (реактивы Гриньяра). Обычно их готовят непосредственным действием магния на соответствующий органический бромид (или иодид) в эфирном растворе, как сказано выше. Из них можно приготовить:
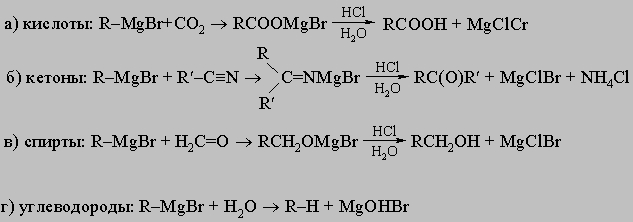
Реакции литийорганических соединений RLi очень похожи на реакции реактивов Гриньяра. Действием реактивов Гриньяра на соответствующие галогениды цинка, кадмия или ртути можно получить как моноалкил- (R-M-Cl), так и диалкилпроизводные (R-M-R ) этих металлов. Из этих соединений цинк- и кадмийалкилы почти так же реакционноспособны, как и реактив Гриньяра, хотя они слабо реагируют с более инертными карбонильными соединениями (кетонами, сложным эфирами). Ртутьалкилы инертны в большинстве реакций, в которые вступают реактивы Гриньяра. Они легко расщепляются только свободными галогенами и сильными неорганическими кислотами. Действием реактивов Гриньяра на хлориды алюминия, олова, германия и свинца можно приготовить частично или полностью алкилированные производные. Из них тетраэтилсвинец имел большое значение как антидетонатор (в этилированном бензине):

2. С0: углеводороды. Парафиновые углеводороды (алканы). Эти соединения, многие из которых встречаются в нефти, соответствуют общей формуле CnH2n + 2. Поскольку в них каждая из связей углерода имеет ковалентный характер, так как замыкается либо на углерод, либо на водород, валентная оболочка углерода полностью насыщена, в результате чего парафиновые углеводороды химически очень инертны. Синтетически парафины могут быть получены восстановлением алкилгалогенидов водородом на таких катализаторах, как палладий на карбонате кальция, действием цинка в спирте или магния (через RMgBr) с последующей обработкой водой:

Их можно также приготовить прямым восстановлением спиртов водородом на медно-хромовых катализаторах при высоких температурах и давлениях, а также действием иодоводорода на спирты при 180° С или гидрированием олефинов и ацетиленов на таких катализаторах, как палладий, платина или никель. Технически важным методом получения низших гомологов, которые представляют собой ценное топливо, является крекинг. В этом процессе высшие гомологи, проходя через нагретую до 500-700° С трубку, расщепляются на более простые соединения, например:
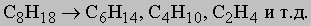
Смесь углеводородов, пригодных в качестве топлива, можно приготовить в промышленных масштабах по Фишеру - Тропшу. В этом процессе смесь СО и H2 (в отношении 1:2) пропускают над кобальтовым или никелевым катализатором при 200° С. Из-за их инертности к большинству химических реагентов углеводороды не представляют большого интереса для синтетической органической химии. На солнечном свету они реагируют с хлором, производя хлорированные углеводороды, например:

Обычно образуются смеси различных возможных продуктов, поэтому реакция имеет ценность главным образом для получения растворителей, где разделение компонентов несущественно. В некоторых случаях посредством фракционной перегонки получают чистые продукты. При высоких температурах парафины реагируют также с азотной кислотой, производя нитропарафины:
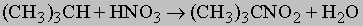
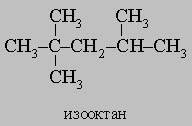
Образующееся вещество, 2-нитро-2-метилпропан, является ценным растворителем. Нормальные углеводороды часто удается изомеризовать в разветвленные углеводороды при действии безводного хлорида алюминия. Эта реакция важна для производства моторных топлив с низкой способностью к детонации. Мерой склонности к детонации (преждевременному воспламенению смеси горючего и воздуха в двигателях внутреннего сгорания) служит октановое число бензина, которое определяют сравнением со стандартными смесями гептана (октановое число 0) и 2,2,4-триметилпентана (т.н. "изооктана", октановое число 100):
Олефины (алкены). Эти соединения, простейшим представителем которых является этилен H2C=CH2, соответствуют общей формуле CnH2n. Они содержат двойную связь

.
Такая связь способна присоединять реакционноспособные атомы и группы, причем каждый из участвующих в ней атомов углерода в результате образует четыре простые связи, поэтому двойная связь называется ненасыщенной. Олефины могут быть получены: а) каталитическим дегидрированием парафинов над оксидом хрома или другими катализаторами
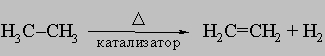
.
б) дегидрацией спиртов в присутствии серной кислоты или оксида алюминия при высоких температурах
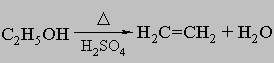
.
в) отщеплением галогеноводорода от алкилгалогенидов при помощи сильных оснований, например этилата калия:
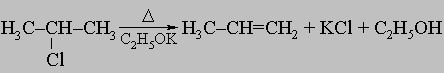
.
(когда может образоваться более одного олефина, преобладает наиболее разветвленный). Олефины вступают в следующие реакции: а) с водородом на платиновых и сходных с ними катализаторах, давая парафины (см. выше); б) с галогенами, давая вицинальные (виц) дигалогениды, в которых атомы галогенов присоединены к двум соседним углеродным атомам:

.
в) с пероксидом водорода, перманганатом калия или тетраоксидом осмия, давая гликоли:
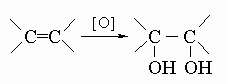
.
г) с серной кислотой, давая алкилсерные кислоты, которые можно прогидролизовать до спирта:

.
д) с галогеноводородами, давая алкилгалогениды:
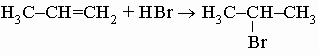
.
е) с хлорноватистой кислотой, давая хлоргидрины:
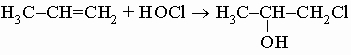
.
(По правилу Марковникова, при присоединении протонных кислот или воды к несимметричным алкенам или алкинам атом водорода присоединяется к наиболее гидрированному атому углерода.) В присутствии свободных радикалов, например радикалов, образующихся при разложении ацетилоксида H3C-C(O)-O-O-C(O)CH3, многие органические соединения гладко присоединяются к олефиновым связям, например,
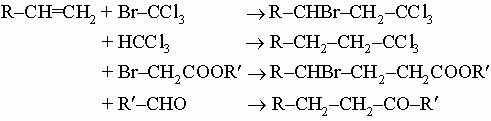
.
Простейший олефин - этилен - применяется как регулятор роста растений, ускоряющий созревание плодов (в том числе цитрусовых), и как исходное соединение в производстве полиэтилена, полиэтилен-пропиленовых каучуков, а также для синтеза этиленгликоля CH2OH-CH2OH. Во время Первой мировой войны его широко применяли для производства иприта (горчичного газа) ClCH2CH2SCH2CH2Cl, который получают действием дихлорида серы на этилен.
См. также ХИМИЧЕСКОЕ И БИОЛОГИЧЕСКОЕ ОРУЖИЕ. Многие диены, т.е. углеводороды, содержащие две двойные связи, представляют промышленный интерес. Бутадиен H2C=CH-CH=CH2, хлоропрен H2C=CCl-CH=CH2 и изопрен H2C=C(CH3)CH=CH2 полимеризуются, давая каучук. Природный каучук также можно рассматривать как полиизопреноиды природного происхождения (см. разд. IV-1.А.4 и IV-2.Б.1).
См. также КАУЧУК И РЕЗИНА.
Ацетилены. Карбид кальция CaC2 при обработке водой выделяет газ ацетилен C2H2, имеющий структуру H-C=C-H. Это вещество является первым членом гомологического ряда ацетиленовых углеводородов CnH2n - 2. Наиболее общий путь получения соединений этого ряда состоит в присоединении брома к соответствующим олефинам с последующей обработкой спиртовым раствором гидроксида калия. Присоединением воды в присутствии сульфата ртути и серной кислоты ацетилен превращается в уксусный альдегид, из которого можно получить уксусную кислоту и другие ценные технические продукты:
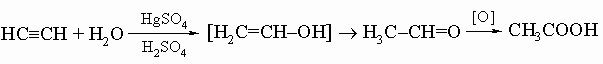
(Соединения со структурой

неустойчивы, так как самопроизвольно перегруппировываются в
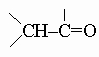
.)
Ацетилен и те из его гомологов, у которых имеется водород при связанном тройной связью углероде, ведут себя как очень слабые кислоты. Их соли со щелочными металлами можно получить действием амида натрия или амида калия:

С аммиачным раствором серебра или одновалентной меди ацетилены образуют нерастворимые взрывчатые соли серебра и одновалентной меди. У всех соединений ряда тройная связь способна присоединять реагенты подобно двойной связи.
3. Окислительное состояние С+. Алкилгалогениды можно рассматривать как производные углеводородов, у которых водород заменен галогеном. Они имеют общую формулу R-X, где X может быть F, Cl, Br или I. Прямое замещение водорода галогеном редко может служить препаративным методом получения алкилгалогенидов (см. выше). Более подходящий метод состоит в обработке соответствующего спирта (ROH) галогеноводородом или галогенидом фосфора, чтобы заменить гидроксильную группу на галоген. По многим физическим свойствам, таким, как низкие температуры кипения и плавления, алкилгалогениды напоминают углеводороды, поскольку оба класса соединений относительно неполярны. Химически алкилгалогениды гораздо более реакционноспособны, причем иодиды наиболее активны, а хлориды - наименее. По реакционной способности, реакциям и методам получения алкилфториды сильно отличаются от других галогенидов. Получение металлалкилов, например реактивов Гриньяра, уже обсуждалось в разд. IV-1.А.1. Атом галогена можно также заменить на самые разнообразные простые неорганические или органические основания, например:

Такие реакции замещения лучше идут с алкилбромидами и алкилиодидами, хлор в алкилхлоридах заменить труднее. Параллельно с написанными выше идут побочные процессы - реакция с растворителем и отщепление HX с образованием олефина. Природа R оказывает сильнейшее влияние на скорость и состав продуктов реакции. Реакции замещения могут протекать по двум различным механизмам: мономолекулярному (SN1) или бимолекулярному (SN2). Согласно первому механизму сначала происходит диссоциация алкилгалогенида на галогенид-анион и ион карбения - нестабильную высокореакционноспособную частицу, которая немедленно реагирует с добавленным основанием или молекулой растворителя. Поскольку стабильность карбениевых ионов растет от первичных к третичным
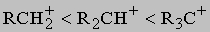
этот механизм замещения должен быть преобладающим для третичных алкилгалогенидов R3CX, его роль должна снижаться для вторичных алкилгалогенидов R2CHX. По второму механизму вступающая группа постепенно вытесняет уходящую, причем в переходном состоянии обе группы связаны с углеродом в реакционном центре приблизительно одинаково. Наиболее энергетически выгодным направлением атаки для вступающей группы является подход со стороны, обратной направлению, в котором удаляется входящая группа:
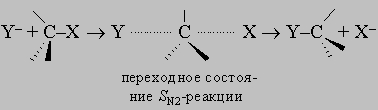
Поскольку в переходном состоянии электронная плотность на реакционном центре выше, чем в исходном и конечном, скорость такого процесса должна падать в ряду RCH2X > R2CHX > R3CX, т.е. в последовательности, обратной той, которая характерна для SN1-реакций.
Реальные процессы замещения являются чем-то промежуточным по отношению к двум описанным крайним идеальным случаям, причем реальный механизм замещения для первичных алкилгалогенидов RCH2X будет близок к SN2, а для третичных алкилгалогенидов R3CX - к SN1, тогда как для вторичных алкилгалогенидов R2CHX реальный механизм будет представлять собой нечто среднее. Поэтому наблюдаемые скорости замещения обычно уменьшаются при переходе от RCH2X к R2CHX. Одновременно при переходе от первичных алкилгалогенидов ко вторичным и третичным возрастает роль упомянутых выше побочных процессов - реакций с растворителем (водой, спиртом и т.п.) и образования олефинов, которые в случае некоторых вторичных и особенно третичных алкилгалогенидов могут стать преобладающими. Присутствие двойной связи вблизи галогена также сильно изменяет реакционную способность. Так, винилгалогениды R-CH=CH-X и арилгалогениды ArX исключительно малоактивны; наоборот, аллилгалогениды R-CH=CH-CH2X и бензилгалогениды Ar-CH2X необычайно реакционноспособны. Все перечисленные выше реагенты являются основаниями Льюиса и могут вызывать конкурирующую реакцию отщепления, в которой отщепляется галогеноводород и образуется олефин:

Эта реакция идет особенно хорошо с такими сильными основаниями, как OH- и RO-, и становится преобладающей, когда используются третичные галогениды или если реагентами являются спиртовые растворы сильных оснований:

Простые полихлорированные углеводороды широко применяются в промышленности в качестве растворителей. Среди наиболее важных растворителей можно упомянуть хлороформ CHCl3, дихлорэтан ClCH2CH2Cl и тетрахлорэтан Cl2CHCHCl2.
Спирты и простые эфиры. Одноатомные спирты имеют общую формулу R-OH, представляющую углеводород, в котором водород заменен гидроксильной группой. Далее они могут быть подразделены на первичные RCH2OH, вторичные RR'CHOH и третичные спирты, RR'R "COH, в зависимости от того, одна, две или три алкильные группы присоединены к углероду, несущему гидроксильную группу. Низшие спирты находят широкое применение в промышленности в качестве растворителей и как промежуточные вещества для синтеза. Метанол (т. кип. 64,7° С) получают взаимодействием CO и H2 при высоком давлении над хромо-цинковым оксидным катализатором при 350-400° С. Этанол (обычный этиловый спирт, т. кип. 78,3° С) традиционно получают сбраживанием сахара или крахмала в присутствии дрожжей, хотя некоторое количество его производят путем поглощения этилена серной кислотой с последующим гидролизом образующейся этилсерной кислоты C2H5OSO3H водой. Оба процесса дают разбавленные спиртовые растворы, из которых получают перегонкой поступающий в продажу 95%-й спирт. Изопропиловый спирт (пропанол-2, т. кип. 82,3° С) обычно делают сернокислотным методом из пропилена CH3CH=CH2, побочного продукта производства бензина крекингом. Он находил некоторое применение как заменитель этанола в качестве растворителя и в спиртовых растираниях. Некоторые из высших спиртов, например 2-этилгексанол-1 (или "612"), действуют на насекомых как репелленты. Общие методы лабораторного получения спиртов включают а) гидролиз алкилгалогенидов; б) гидратацию олефинов в присутствии минеральных кислот, например описанным выше сернокислотным методом; в) действие реактивов Гриньяра RMgX на альдегиды R'-CHO и кетоны R'-CO-R ". Формальдегид дает первичные спирты, альдегиды - вторичные спирты, а кетоны - третичные:

Спирты обнаруживают свойства очень слабых кислот. Водород гидроксильной группы в спиртах несколько менее кислый, чем водород воды. Он может быть замещен на активные металлы с образованием алкоголятов:

Эта реакция легче всего протекает с первичными спиртами и медленнее - с третичными. Na реагирует очень медленно с трет-бутиловым спиртом, но K (более активный) реагирует быстро. Вообще реакции спиртов, в которых рвется O-H-связь, легче всего протекают с первичными спиртами и медленнее всего - с третичными. Сложные эфиры можно получить следующим образом:

Образование сложных эфиров по первым двум из этих реакций идет быстро и необратимо и, как правило, не требует катализаторов (хотя обычно к реакционной смеси прибавляют такие основания, как пиридин или триэтиламин, которые связывают образующиеся кислоты в виде солей). Третий метод основан на обратимой равновесной реакции и требует катализатора, обычно кислоты (этерификация по Фишеру). Так, реакцию можно заставить протекать слева направо (гидролиз), если использовать избыток спирта и удалять воду по мере ее образования. Ни один из трех указанных выше методов не применим к третичным спиртам. Спирты, однако, амфотерны и в присутствии сильных кислот ведут себя как очень слабые основания:

Способность к замещению группы -OH в этой и других реакциях убывает от третичных к первичным спиртам. В присутствии таких дегидратирующих агентов, как серная или фосфорная кислота, при более низких температурах из спиртов образуются простые эфиры R-O-R, тогда как при более высоких температурах путем отщепления воды получаются олефины. Этот метод не годится для получения простых эфиров из вторичных спиртов, а с третичными дает только олефины. Дегидратация спиртов с образованием олефинов может быть осуществлена каталитически в паровой фазе над такими оксидами металлов, как оксид алюминия. Окисление спиртов можно осуществить при помощи сильных окислителей (хромовая или азотная кислота). Продукты окисления различны по своей природе для первичных, вторичных и третичных спиртов. Так, первичные спирты сначала окисляются в альдегиды, которые, если их немедленно не удалить из окислительной среды, окисляются далее до кислот:

Вторичные спирты окисляются до устойчивых кетонов RCOR', тогда как третичные спирты окисляются только очень сильными окислителями, расщепляющими молекулу на кислоты и кетоны с меньшим числом углеродных атомов. Амины можно рассматривать как производные аммиака, получаемые последовательным замещением атомов водорода алкильными группами, аналогично тому, как спирты и просто эфиры можно представить как производные воды. В соответствии с числом замещенных атомов водорода различают первичные (R-NH2), вторичные (RR'NH) и третичные амины (RR'R "N). Дальнейшее алкилирование дает четвертичные аммониевые соли RR'R "R "'N+X-, которые можно рассматривать как полностью алкилированную аммониевую соль NH4+X-. Прямое алкилирование аммиака алкилгалогенидами имеет очень ограниченную ценность для получения первичных аминов, поскольку эту реакцию трудно контролировать и она ведет к смесям первичных, вторичных и третичных продуктов. Прямое алкилирование первичных аминов, однако, часто используют для получения вторичных и третичных аминов, а также четвертичных аммониевых солей. Для приготовления первичных аминов существует много хороших альтернативных методов, например: 1) расщепление амидов щелочным гипобромитом или гипохлоритом (реакция Гофмана). Промежуточными соединениями в этой реакции являются изоцианаты, и этот метод можно использовать для их получения:

2) перегруппировка азидов кислот в изоцианаты (см. разд. IV-1.А.6) с последующим их гидролизом до аминов (реакция Курциуса):

3) реакция алкилгалогенидов с фталимидом калия с последующим гидролизом продукта (реакция Габриэля):

4) восстановление нитрилов натрием в спирте или водородом на никелевом катализаторе под давлением в присутствии аммиака:

Другие полезные методики этого рода включают восстановление нитросоединений R-NO2, оксимов R-CH=NOH и гидразонов R-CH=N-NH2. Амины - несколько более сильные основания, чем аммиак; константы диссоциации для реакции
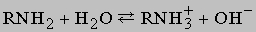
составляют примерно 10-4. Поэтому амины легко образуют с большинством кислот соли, подобные солям аммония. Первичные амины реагируют с азотистой кислотой, давая спирты

но реакция является сложной и ведет также к олефинам и спиртам, изомерным с ожидаемыми. При взаимодействии с нитрозилбромидом из аминов образуются алкилбромиды
 .
.
Бензоилированные вторичные амины (R-NH-COC6H5) при перегонке с пентабромидом фосфора также превращаются в алкилбромиды (реакция Брауна). Вторичные амины дают с азотистой кислотой N-нитрозопроизводные RR'N-N=O, тогда как третичные амины просто образуют соли. Это заметное различие в поведении трех типов аминов по отношению к азотистой кислоте часто используется, чтобы отличить их друг от друга. Четвертичные аммониевые соли образуются при действии алкилгалогенидов на третичные амины. Обработка оксидом серебра превращает эти соли в соответствующие гидроксиды, которые являются основаниями, сравнимыми по силе с гидроксидами натрия и калия. При нагревании они разлагаются на амины и олефины с выделением воды. Такое поведение лежит в основе расщепления по Гофману, широко использовавшегося при установлении структуры алкалоидов для определения положения азота в цепи или гетероциклическом кольце. Амин (например, 3-аминопентан) исчерпывающим метилированием (обработка избытком метилиодида и сильным основанием) превращают в иодид триметилалкиламмония, например (C2H5)2CH-N+(CH3)3I-, и затем действием оксида серебра - в соответствующий гидроксид, например (C2H5)2CH-N+(CH3)3OH-. Последний при перегонке дает воду, триметиламин и олефин, например CH3CH=CHCH2CH3. Исходное положение аминогруппы можно вывести из положения двойной связи. Первичные и вторичные (но не третичные) амины реагируют с кислотами при нагревании, образуя амиды,

но эта реакция обратима и чаще амиды получают из аминов и хлорангидридов или ангидридов (см. разд. IV-1.А.5). С бензолсульфохлоридом первичные и вторичные амины дают бензолсульфамиды (третичные амины опять-таки не реагируют по этой схеме):
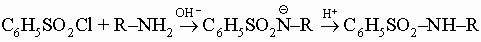
Сульфамиды первичных аминов растворимы в основаниях, а сульфамиды вторичных аминов нейтральны; таким образом, эта реакция может быть использована для разделения трех классов аминов (разделение по Гинзбергу).
Нитросоединения. Эти соединения имеют общую формулу R-NO2. Их можно получать взаимодействием алкилгалогенидов с нитритом серебра или натрия, а также взаимодействием углеводородов с азотной кислотой. Вторичные и третичные водороды удается заместить нитрогруппами при нагревании разветвленных углеводородов с концентрированной азотной кислотой в запаянной трубке при 150° С. Промышленное получение нитросоединений часто осуществляют в паровой фазе при 400-500° С. В промышленности нитросоединения используют как растворители и как исходные соединения для получения алифатических аминов (см. выше, методы получения первичных аминов, п. 4). Меркаптаны, тиоэфиры и получаемые из них сернистые производные. Меркаптаны (тиоспирты) R-SH свое название получили за легкость, с которой они образуют характерные нерастворимые ртутные соли (corpus mercurio aptum, т.е. тела, связывающие ртуть). Меркаптаны получают действием гидросульфида калия на алкилгалогениды или взаимодействием спиртов с сероводородом в паровой фазе при 300-350° С над диоксидом тория. Они являются летучими жидкостями с неприятным запахом и несколько более низкими температурами кипения, чем соответствующие спирты. Подобно сероводороду, они представляют собой слабые кислоты (немного сильнее, чем спирты) и легко образуют устойчивые металлические соли, или меркаптиды, например R-SNa или (R-S)2Pb. Мягкие окислители, включая кислород воздуха, превращают их в диалкилдисульфиды R-S-S-R, из которых их можно регенерировать посредством самых различных восстановителей. С альдегидами в присутствии хлороводорода или других кислых катализаторов они дают меркаптаны, например R'CH(SR)2. Тиоэфиры R-S-R' можно получить взаимодействием алкилгалогенидов с сульфидом калия (2RI + K2S -> R-S-R' + 2KI) или с меркаптидами калия (RI + R'SK -> R-S-R' + KI). Это летучие жидкости без неприятного запаха, кипящие несколько выше, чем соответствующие меркаптаны. С алкилгалогенидами они дают устойчивые кристаллические соли сульфония RR'R "S+X-, которые по своим свойствами напоминают соли аммония. Сульфониевые основания RR'R "S+OH- сравнимы по силе с едким натром. Окисление тиоэфиров через неусточивые сульфоксиды RR'SO ведет к стабильным сульфонам RR'SO2. Окисление меркаптанов под действием HNO3, KMnO4 или H2O2 ведет в алкилсульфоновым кислотам R-SO3H. Эти соединения можно также получить прямым сульфированием углеводородов (RH + H2SO4 -> RSO3H + H2O) или взаимодействием сульфитов металлов с алкилгалогенидами. Они являются сильными кислотами, легко растворимы в воде и дают кристаллические соли (R-SO3M).
4. С2+: альдегиды и кетоны. Многие альдегиды и кетоны, особенно альдегиды и кетоны терпенового ряда (терпены - ненасыщенные углеводороды, получающиеся димеризацией изопрена CH2=C(CH3)-CH=CH2), встречаются в природе и имеют большое значение в парфюмерной промышленности. Вещества этого рода включают цитронеллаль (CH3)2C=CHCH2CH2CH(CH3)CH2CHO из масла цитронеллы и эвкалипта и цитраль (CH3)2C=CHCH2CH2C(CH3)=CHCHO из масла цитронеллы.
Получение. Формальдегид (т. кип. -21° С) получают каталитическим дегидрированием метанола над нагретым серебряным или медным катализатором. Водный 40%-й раствор формальдегида продается под названием формалин. Ацетальдегид получают каталитической гидратацией ацетилена. Ацетальдегид является исходным веществом для получения уксусной кислоты CH3COOH. Ацетон CH3COCH3 (пропанон), простейший кетон, представляет собой ценный растворитель. Его получают пропусканием паров уксусной кислоты над нагретым оксидом марганца:

Общие методы получения альдегидов включают: а) окисление первичных спиртов хромовой или надсерной кислотами в присутствии иона двухвалентного железа, причем первый метод подходит только для получения летучих альдегидов, которые можно немедленно отогнать из реакционной смеси прежде, чем произойдет дальнейшее окисление; б) пиролиз кальциевой соли карбоновой кислоты с избытком муравьинокислого кальция:
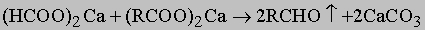
и в) восстановление хлорангидридов RCOCl водородом над слегка дезактивированным палладием на сульфате бария (реакция Розенмунда). Кетоны можно получить а) окислением вторичных спиртов хромовой кислотой при низкой температуре

б) пиролизом кальциевых, бариевых и ториевых солей карбоновых кислот

в) действием алкилцинк- или алкилкадмийгалогенидов на ацилгалогениды

г) действием реактивов Гриньяра на нитрилы
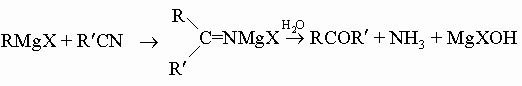
Окисление альдегидов до кислот лучше всего осуществлять при низкой температуре, используя а) перманганат калия KMnO4 в воде или ацетоне; б) надкислоты, например надуксусную кислоту CH3C(O)-O-OH, или в) аммиачный раствор нитрата серебра. Последний реагент полезен, если нужно отличить альдегиды от кетонов, поскольку небольшое количество альдегида вызывает образование красивого налета "серебряного зеркала" на стенках реакционного сосуда:

Окисление кетонов редко используется в синтетических целях. Особый тип окисления, известный как галоформная реакция, претерпевают ацетальдегид и метилкетоны (а также спирты, которые могут быть окислены в эти соединения) при действии гипогалогенитов (растворы галогенов в едком натре):
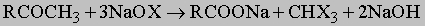
Когда используется иод, образуется объемистый желтый осадок иодоформа CHI3. Эту реакцию применяют, чтобы отличить кетоны от их высших гомологов. Восстановление альдегидов в первичные спирты осуществляется каталитическим гидрированием при комнатной температуре над платиной в присутствии следов двухвалентного железа. Восстановления кетонов во вторичные спирты можно добиться подобным же образом над платиной, хромитом меди или никелем, хотя приходится использовать несколько более высокие давления водорода. Альдегиды и кетоны можно восстановить в спирты литийалюминийгидридом LiAlH4 или борогидридом натрия NaBH4. Карбонильную группу можно восстановить до метиленовой по Клемменсену (амальгамой цинка в соляной кислоте) или по Кижнеру - Вольфу (обработка гидразона горячей щелочью).
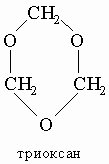
Полимеризация. Альдегиды склонны полимеризоваться, особенно при контакте с воздухом или в присутствии кислотных либо основных катализаторов. При кислотном катализе полимеризация формальдегида ведет к образованию тримера - триоксана (или a-триоксиметилена)
а также к полимерам с прямой цепью различной длины, называемым полиоксиметиленами, HO-CH2-(OCH2)n-O-CH2OH, содержащим до 100 формальдегидных единиц. Триоксан представляет собой удобную твердую модификацию формальдегида, поскольку из него можно регенерировать газообразный мономер просто нагреванием. В присутствии гидроксида кальция формальдегид может давать смесь оксиальдегидов и оксикетонов общей формулы (CH2O)n, из которой были выделены вещества, напоминающие сахара. Аналогичным образом ацетальдегид претерпевает кислотно-катализируемую тримеризацию в паральдегид

а при низких температурах - тетрамеризацию в метальдегид. Альдольная конденсация - это реакция димеризации, которую претерпевают альдегиды и (несколько труднее) кетоны, имеющие водород при углеводородном атоме, соседнем с карбонильной группой. Так, слабые основания вызывают обратимую димеризацию ацетальдегида в альдоль, CH3-CHOH-CH2CHO, а ацетона - в диацетоновый спирт (CH3)2C(OH)CH2COCH3. Кислоты вызывают такую же конденсацию, но далее катализируют отщепление воды, ведущее к ненасыщенным альдегидам и кетонам, которые в свою очередь могут претерпевать дальнейшую конденсацию:

Реакции присоединения по карбонилу. Карбонильная группа является ненасыщенной и потому вступает в многочисленные реакции присоединения. Так (см. разд. IV-1.А.1), получение спиртов легко осуществляется присоединением реактивов Гриньяра по карбонильной группе. Присоединение синильной кислоты ведет к образованию циангидринов (реакция Килиани)
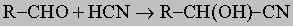
,
а в присутствии хлорида аммония - аминонитрилов R-CH(NH2)-CN. Большое число азотистых оснований общего типа A-NH2 дает при присоединении к карбонильной группе с последующим отщеплением воды кристаллические производные типа A-N=CHR и A-N=CRR'. Так, гидроксиламин NH2OH дает оксимы RCH=NOH; гидразин NH2-NH2 - гидразоны RCH=N-NH2, а семикарбазид H2N-NH-CO-NH2 - семикарбазоны RCH=N-NHCONH2, вещества, ценные для очистки и идентификации карбонильных соединений. Бисульфит натрия присоединяется аналогичным образом к альдегидам и метилкетонам, давая кристаллические водорастворимые производные R-CH(OH)-SO3Na, из которых карбонильные соединения можно регенерировать действием карбоната натрия.
Ацетали. Альдегиды легко реагируют со спиртами в присутствии кислотных катализаторов, образуя ацетали:
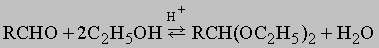
Реакция обратима, альдегиды и спирты легко регенерировать из ацеталей действием воды и кислот; образование ацеталей часто используют, чтобы временно защитить чувствительную альдегидную группу, пока в другой части молекулы выполняются химические превращения. Кетоны обычно не столь легко вступают в эту реакцию, но ацетон конденсируется с полигидроксисоединениями (например, сахарами), образуя циклические ацетали типа

Галогенирование. Карбонильные соединения, имеющие водород при углероде, соседнем с карбонильной группой, легко реагируют с галогенами, давая a-галогенальдегиды или кетоны. Реакция катализируется кислотами и основаниями. Кислотно-катализируемое галогенирование несколько легче контролировать, и потому его часто используют, если желательно ввести только один атом галогена:
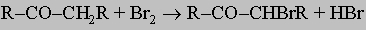
Атом галогена, соседний с карбонильной группой, исключительно активен во многих реакциях замещения. Часто его можно также отщепить вместе с соседним водородом при действии оснований, что дает ненасыщенные карбонильные соединения R-C(O)-CH=CHR'.
5. С3+: карбоновые кислоты и их производные. Нахождение в природе. В природе карбоновые кислоты редко находятся в свободном виде, хотя ожоги крапивой и укусы муравьев и пчел болезненны из-за раздражающего действия свободной муравьиной кислоты. Среди других карбоновых кислот, встречающихся в природе в свободном виде, - изовалериановая (корень валерианы) и изомасляная. В природе карбоновые кислоты встречаются главным образом в виде эфиров и соединений со спиртами. Первые три члена гомологического ряда карбоновых кислот являются относительно высококипящими жидкостями с резким, раздражающим запахом. Высшие члены ряда представляют собой жидкости или низкоплавкие твердые вещества, причем более летучие кислоты обладают крайне неприятным запахом.
Получение. В промышленности натриевую соль муравьиной кислоты получают взаимодействием монооксида углерода с едким натром при 200° С под давлением. При осторожном нагревании этой соли с рассчитанным количеством серной кислоты удается отогнать муравьиную кислоту. Уксусную кислоту получают в промышленных масштабах главным образом окислением уксусного альдегида, получаемого из ацетилена. Водный 5% раствор (столовый уксус) можно приготовить разбавлением промышленной уксусной кислоты или сбраживанием разбавленных растворов спирта уксуснокислыми бактериями. Общие методы получения кислот в лаборатории: а) окисление первичных спиртов азотной кислотой, перманганатом калия или хромовой кислотой
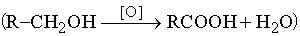
;
б) обработка реактивов Гриньяра углекислым газом
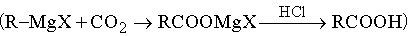
;
в) взаимодействие алкилгалогенидов с цианидом натрия с последующим гидролизом образующихся нитрилов

;
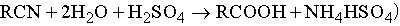
;
г) окислительное расщепление олефинов перманганатом калия
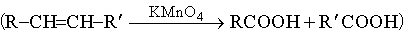
.
В неорганической химии имеется правило, согласно которому окисление элемента, несущего гидроксильную группу, усиливает кислотные свойства этой группы (ср. H2SO4 и H2SO3, HNO3 и HNO2). Аналогично в органической химии карбоновые кислоты проявляют значительно более кислотные свойства, чем спирты (RCOOH и RCH2OH). Константы диссоциации для равновесия RCOOHRCOO- + H+ обычно имеют величину 10-5-10-4 (для спиртов - 10-18). Ввиду достаточно высокой кислотности карбоновых кислот они легко растворяются в бикарбонате натрия, соде или едком натре с образованием натриевых солей (RCOONa). Из них карбоновые кислоты можно выделить действием более сильных минеральных кислот (HCl, H2SO4). Со спиртами карбоновые кислоты реагируют, давая сложные эфиры (RCOOH + R'OHRCOOR' + H2O). Прямая реакция идет медленно, но катализируется сильными неорганическими кислотами. Она обратима, поэтому успешное приготовление сложного эфира из кислоты может быть достигнуто только смещением равновесия путем использования большого избытка спирта или удаления воды из реакционной смеси. Из кислоты можно получить метиловые эфиры в очень мягких условиях действием диазометана:
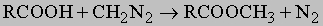
Карбоновые кислоты реагируют с трихлоридом фосфора, пентахлоридом фосфора или тионилхлоридом (SOCl2) с образованием высокореакционноспособных хлорангидридов RCOCl:

Эти очень легко гидролизуемые вещества особенно полезны для получения производных кислот. Так, со спиртами они дают эфиры:

с аммиаком или аминами - амиды:
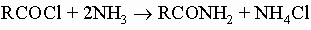
с натриевыми солями карбоновых кислот - ангидриды:

Соли кислот вступают в некоторые полезные реакции. Так, натриевые соли при сплавлении с едким натром дают насыщенные углеводороды RH. При электролизе получаются углеводороды RR. Перегонка кальциевых или ториевых солей приводит к образованию кетонов RCOR. Серебряные соли RCOOAg при обработке алкилгалогенидами R'Br дают эфиры RCOOR', а при действии галогенов - алкилгалогениды:
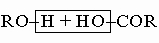
.
Эфиры: жиры и воски. Эти соединения, препаративные методы получения которых обсуждены выше, можно рассматривать как образующиеся из кислоты и спирта при отщеплении воды: . Летучие эфиры, имеющие низкие молекулярные массы, являются душистыми маслами. Многие из них, например этилформиат, этилацетат, изоамилацетат, этилбутират и т.д., обнаружены в душистых цветах и плодах. Наиболее важными среди эфиров с высокой молекулярной массой являются жиры и воски, которые представляют собой масла или низкоплавкие твердые вещества.
Жиры - это триэфиры глицерина (см. ниже) и высших жирных кислот:

Жирные кислоты из природных жиров содержат четное число углеродных атомов в молекуле, причем самыми распространенными являются С16- и С18-кислоты. Кроме насыщенных кислот, в жирах присутствуют многочисленные ненасыщенные кислоты. Высоконенасыщенные жиры - жидкости (растительные и рыбные жиры), тогда как более насыщенные жиры (жиры животных) - твердые вещества. Твердые кулинарные жиры можно получить из жидких жиров частичным гидрированием водородом на никелевом катализаторе. Наиболее обычными из ненасыщенных кислот являются

Глицериды, как и все эфиры, гидролизуются (омыляются) водными щелочами с образованием спирта (глицерина) и соли щелочных металлов входящих в их состав кислот. Соли высших кислот называются мылами. Процесс омыления имеет большое промышленное значение. Во время войн глицерин пользовался большим спросом для получения взрывчатых веществ (см. разд. IV-1.Б.1). Воски являются сложными эфирами высших жирных кислот и высших спиртов. Примерами могут служить пчелиный воск (мирицилпальмитат C15H31COOC31H63) и карнаубский воск (церилцеротат C25H51COOC26H53). Кроме реакций омыления, обсуждавшихся выше, эфиры вступают в общие для них реакции сольволиза с водой, спиртами или аммиаком:
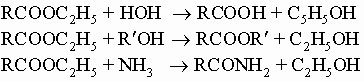
Первые две реакции катализируются неорганическими кислотами. Таким образом, данный сложный эфир при правильном выборе сольволитического агента можно превратить в свободную кислоту, другой эфир или амид. Некоторое препаративное значение имеет реакция восстановления сложных эфиров в спирты. Это может быть осуществлено действием натрия в спирте или гидрированием под давлением при 250° С в присутствии хромита меди. Последний метод используется в промышленности для получения многих малодоступных ранее высших спиртов. Ортоэфиры RC(OR')3, как и ацетали, вполне устойчивы в щелочных растворах, но легко превращаются в сложные эфиры при действии водных кислот. Трихлорметильные соединения RCCl3 можно прогидролизовать до кислот нагреванием с водной щелочью. Сложные эфиры конденсируются в b-кетоэфиры в присутствии этилата натрия C2H5ONa или сходных с ним сильных оснований (см. разд. IV-1.Б.5). Другие производные кислот: амиды и ангидриды. Амиды RCONH2 получают в основном действием аммиака на хлорангидриды RCOCl или на сложные эфиры RCOOR'. Они имеют значение в органическом синтезе, поскольку из них можно приготовить первичные амины RNH2 по реакции Гофмана (разд. IV-1.А.3). При дегидратации фосфорным ангидридом из них образуются нитрилы R-CєN. Ангидриды RC(O)OC(O)R обычно получают действием хлорангидридов на натриевые соли кислот. Они используются в основном для ацилирования спиртов. Так, действием уксусного ангидрида на спирты получают ацетаты:

Кислоты, сложные эфиры, хлорангидриды и ангидриды можно восстановить до спиртов литийалюминийгидридом.
6. С4+: производные угольной кислоты. Эфиры угольной и хлоругольной кислот. При взаимодействии фосгена COCl2 со спиртами первоначально образуются эфиры хлоругольной кислоты R-O-COCl. Эти вещества представляют собой летучие жидкости с острым запахом, применяемые для введения этерифицированной карбоксильной группы в спирты, амины и другие соединения, имеющие активный замещаемый водород. Дальнейшее взаимодействие хлоркарбонатов со спиртами дает карбонаты RO-CO-OR' - летучие жидкости с общими для эфиров свойствами.
Ксантаты. Взаимодействие сероуглерода со спиртами в присутствии щелочей ведет к образованию ксантатов - сложноэфирных солей эфиров дитиоугольной кислоты:

Это кристаллические вещества, которые при обработке кислотами снова выделяют спирты. Применение этой реакции к целлюлозе приводит к получению вискозы - коллоидного раствора ксантата целлюлозы
который при выдавливании в кислый раствор дает красивые нити регенерированной целлюлозы или вискозного шелка.
Производные карбаминовой кислоты. Карбаминовая (моноамидугольная) кислота H2NCOOH известна только в форме солей, сложных эфиров и амидов. Ее эфиры H2NCOOR (уретаны) - кристаллические вещества. Многие из них используются в медицине как седативные средства, например апонал H2NCOOC(CH3)2C2H5 и гедонал H2NCOOCH(CH3)C3H7. Они могут быть получены взаимодействием а) аммиака и эфиров хлоругольной кислоты, б) карбамилхлорида ClCONH2 и спиртов, в) мочевины и спиртов. Замещенные по азоту уретаны получают действием спиртов на изоцианаты

Изоцианаты получают по реакции Гофмана или Курциуса. Мочевину NH2CONH2 можно рассматривать как диамид угольной кислоты или амид карбаминовой кислоты. У млекопитающих она выделяется как главный конечный продукт азотного обмена. Взрослый человек выделяет в день 20-30 г мочевины. Историческое значение мочевины состоит в том, что ее синтезом из цианата аммония Велер в 1828 показал отсутствие мистического барьера между органической и неорганической химией. Синтез Велера еще используется как источник этого соединения:

хотя более новый процесс, состоящий в нагревании карбамата аммония под давлением, уже заменил его в промышленности:
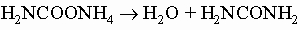
Мочевину получают также взаимодействием цианамида NH2CN с водой. Замещенные мочевины можно синтезировать действием аминов на фосген или изоцианаты:

Мочевина имеет промышленное значение как удобрение и как промежуточное вещество в получении снотворных и успокоительных средств типа
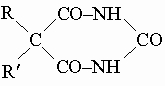
например веронала (R = R' = C2H5), люминала (R = C6H5, R' = C2H5) и пропанала (R = R' = C3H7). Гуанидин HN=C(NH2)2 - имидопроизводное мочевины; его тиоцианат можно получить нагреванием тиоцианата аммония NH4SCH до 150° С. Гуанидин образуется также при взаимодействии аммиака с цианамидом. Это бесцветное кристаллическое гигроскопичное вещество с сильными основными свойствами. Тиомочевина S=C(NH2)2 - сернистый аналог мочевины - может быть получена перегруппировкой тиоцианата аммония в его точке плавления, 149° С. Тиомочевина легко реагирует с алкилгалогенидами, давая кристаллические алкилтиурониевые соли [[RS-(NH2)C=NH2+]]X-. Производные циановой и тиоциановой кислот. Алкилизоцианаты R-N=C=O - промежуточные продукты в реакции Гофмана (превращение амидов карбоновых кислот в первичные амины под действием гипогалогенитов щелочных металлов) и реакции Курциуса (получение первичных аминов термической перегруппировкой ацилазидов в изоцианаты с последующим их гидролизом). Их можно также получить взаимодействием гидрохлоридов аминов (RNH3Cl) и фосгена. Это летучие жидкости с острым запахом, которые легко тримеризуются в циануровую кислоту

При взаимодействии со спиртами они дают уретаны (см. выше), а при кислотном или щелочном гидролизе - амины. Сернистые аналоги этих соединений - изотиоцианаты - летучие жидкости, известные как горчичные масла (это название связано с их острым запахом). Многие из них встречаются в природе в виде гликозидов (см. ГЛИКОЗИДЫ). Так, аллилгорчичное масло CH2=CHCH2N=C=S находится в виде гликозида в черной горчице; этот гликозид может быть расщеплен под действием кислот или фермента мирозина:

Б. ПОЛИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
В органической химии существуют неограниченные возможности для получения соединений, имеющих несколько различных функциональных групп (например, -OH, -COOH, -C(=O)-) в одной и той же молекуле. В общем случае, если такие группы достаточно удалены друг от друга, можно считать, что они будут обнаруживать поведение и реакции, характерные для каждой из них в отдельности. Однако, если они расположены близко друг к другу, возникают классы соединений, требующие особого рассмотрения. Ниже приведены некоторые наиболее важные примеры.
1. Многоатомные спирты и их производные. Гликоли. Простейшим многоатомным спиртом является двухатомный (т.е. две OH-группы) вицинальный (т.е. OH-группы находятся при соседних атомах С) спирт этиленгликоль CH2OH-CH2OH (этандиол), который может быть получен из этиленхлоргидрина CH2OH-CH2Cl гидролизом в присутствии карбоната натрия. Этиленгликоль находит широкое использование как антифриз, а его моноалкиловые эфиры RO-CH2CH2OH, известные под названием целлозольвы, широко применяются как растворители для полиролей и лаков. Высшие гомологи этиленгликоля включают гликоли R-CHOH-CHOH-R', семипинаконы (RR'COH-CHOH-R ") и пинаконы (RR'COH-COHR "R "'). Общие методы их получения включают гидролиз соответствующих вицинальных дигалогенидов (RCHBr-CHBrR') или галогенгидринов (RCHOH-CHBrR'), которые образуются (см. разд. IV-1.А.2) присоединением галогенов или гипогалогеновых кислот (HOX) к олефинам; олефиновая двойная связь может быть также непосредственно окислена холодным разбавленным перманганатом калия, тетраоксидом осмия, пероксидом водорода или его ацилпроизводными, например надуксусной кислотой CH3-C(O)-O-OH. В дополнение к обычным реакциям спиртов гликоли особенно чувствительны к окислительному расщеплению. Так, они количественно окисляются до альдегидов иодной кислотой или тетраацетатом свинца:

Семипинаконы и пинаконы расщепляются аналогичным образом. Пинаконы особенно чувствительны к действию неорганических кислот, в присутствии которых происходит их молекулярная перегруппировка в пинаколины:

Глицерин CH2OH-CHOH-CH2OH - простейший трехатомный спирт, промежуточный продукт в производстве мыла, образуется при омылении жиров (см. разд. IV-1.А.5). Его триэфир азотной кислоты CH2NO3-CHNO3-CH2NO3 - взрывчатое вещество нитроглицерин - образуется при обработке глицерина смесью азотной и серной кислот. Нитроглицерин взрывается от малейшего сотрясения, но адсорбция на кизельгуре (диатомит) повышает его устойчивость; в таком виде он известен как динамит. Трехатомные спирты, как и гликоли, подвержены окислительному расщеплению иодной кислотой:

2. Гидроксиальдегиды и кетоны.
a-Гидроксикарбонильные соединения
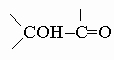
могут быть получены действием бикарбоната натрия на соответствующие a-галогеноангидриды и кетоны

.
a-Кетоспирты типа R-CO-CH2OH получаются кислотно-катализируемым гидролизом диазокетонов R-CO-CHN2, которые могут быть получены действием диазометана CH2N2 на ацилхлориды (хлорангидриды карбоновых кислот). При взаимодействии a-кетоспиртов с тетраацетатом свинца образуются их ацетаты: a-Гидроксикарбонильная группировка часто встречается в природных продуктах, она свойственна сахарам, например альдозам HOCH2-(CHOH)n-CHO (n = 2 - 4) и кетозам CH2OH-(CHOH)n-CO-CH2OH (n = 2, 3). Она легко окисляется; так, под действием иодной кислоты и тетрацетата свинца a-кетоспирты подвергаются мягкому расщеплению:
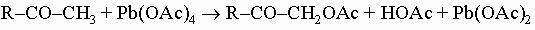
В концевом положении, например -CHOH-CHO или -COCH2OH, группировка взаимодействует с аммиачным раствором нитрата серебра, давая серебряное зеркало, и с ионом меди в щелочном растворе тартрата (фелингова жидкость) или цитрата (реактив Бенедикта), образуя красный осадок оксида меди(I), или медное зеркало, а также с фенилгидразином C6H5NHNH2, давая озазоны (C6H5)HNN=C(R)-CH=N-NHC6H5, которые можно гидролизовать в соответствующие a-кетоальдегиды (дикарбонильные соединения).
b-Гидроксикарбонильные соединения. Когда ацетальдегид обрабатывают мягким основанием (например, карбонатом калия), происходит его димеризация в альдоль (b-гидроксиальдегид) CH3CHOHCH2CHO. Аналогично ацетон может димеризоваться в диацетоновой спирт (CH3)2C(OH)CH2C(O)CH3 (b-гидроксикетон). Эта реакция представляет собой метод получения b-гидроксикарбонильных соединений, или альдолей, и называется альдольной конденсацией. Реакция обратима и может осуществляться только при наличии хотя бы у одного реагента атома H в a-положении к карбонильной группе. Альдольная конденсация двух различных карбонильных соединений ведет к сложным смесям. Формальдегид неспособен к димеризации, поэтому особым синтетическим приложением этого метода является соальдолизация кетонов и альдегидов с формальдегидом, например:

Сходная конденсация катализируется кислотами, но в этих условиях альдоль обычно дегидратируется в соответствующий a,b-ненасыщенный альдегид или кетон, например,
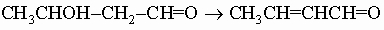
3. Дикарбонильные соединения. Эти вещества имеют общую формулу R-CO-(CH2)nCO-R' и подразделяются на диальдегиды (R = R' = H, алкандиали), кетоальдегиды (R = H, алканалоны) и дикетоны (алкандионы). Они обозначаются как a, b, g, ... дикарбонильные соединения в соответствии со значениями n = 0, 1, 2, ... .
a-Дикарбонильные соединения. Их простейший представитель - глиоксаль CHO-CHO (этандиаль), желто-зеленое летучее вещество, получаемое окислением ацетальдегида азотной кислотой. Глиоксаль легко полимеризуется. В щелочном растворе он превращается в гликолевую кислоту HOCH2COOH. При действии аммиака и формальдегида он дает гетероциклическое соединение имидазол (см. разд. IV-4.Г). Монозамещенные глиоксали (например, метилглиоксаль, или пропаналон-2) можно получить гидролизом соответствующих изонитрозокетонов (R-CO-CH=NOH), которые образуются при взаимодействии алкилнитритов (R-O-N=O) с кетонами в присутствии этилата натрия. Как и глиоксаль, эти соединения склонны к полимеризации и в щелочных растворах превращаются в соответствующие a-гидроксикислоты. Простейшим a-дикетоном является диацетил CH3COCOCH3 (бутан-2,3-дион), желтая летучая жидкость, найденная в некоторых эфирных маслах, например эфирном масле тмина. Его диоксим (диметилглиоксим) используется в аналитической химии для качественного и количественного определения иона никеля. a-Дикетоны можно получать из кетонов путем превращения последних в изонитрозокетоны:

с последующим гидролизом изонитрозокетонов в кислой среде:
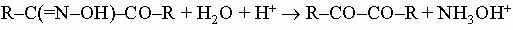
a-Дикетоны можно также получить конденсацией Клайзена из эфиров щавелевой кислоты:

a-Дикетоны являются полезными промежуточными соединениями в синтезе некоторых гетероциклических веществ (см. ниже). Так, при действии аммиака и альдегида из них образуются производные имидазола. Обработка a-диаминами дает пиперазины.
b-Дикетоны. Самый общий путь получения этих соединений - клайзеновская конденсация сложных эфиров с кетонами:

b-Дикетоны представляют собой слабые кислоты, поскольку атомы водорода метиленовой группы сильно активируются соседними карбонильными группами. Натриевые соли дикетонов при обработке алкилгалогенидами дают алкилзамещенные b-дикетоны:

Простейший член ряда - ацетилацетон (пентан-2,4-дион) легко образует комплексные хелатные соли со многими металлами, например,
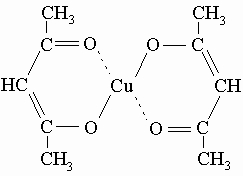
Такие соли обычно интенсивно окрашены, растворимы в хлороформе и других неполярных растворителях, некоторые из них (соли бериллия, алюминия) летучи. b-Дикетоны чувствительны к действию водных щелочей, которые расщепляют их на кетоны и кислоты. Их часто используют как промежуточные соединения в синтезе пиразолов. g-Дикетоны можно получить действием алкилцинкгалогенидов на сукцинилхлорид:
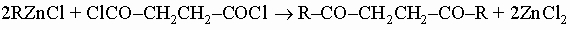
или из b-кетоэфиров:
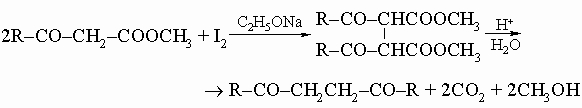
Они полезны для синтеза некоторых гетероциклических соединений (см. разд. IV). 4. Кислоты, содержащие еще одну функциональную (некарбоксильную) группу. Галогенокислоты. Из галогенокислот только a-галогенокислоты R-CHX-COOH можно получить прямым галогенированием, обычно обработкой кислот или соответствующих хлорангидридов (RCH2COCl) хлором или бромом в присутствии красного фосфора. Образующиеся a-галогенацилгалогениды легко гидролизуются до свободных кислот. b-Бромкислоты обычно готовят присоединением бромоводорода к a,b-ненасыщенной кислоте:
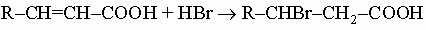
Галогенокислоты с атомом галогена, более удаленным от карбоксильной группы, получают действием бромо- или хлороводорода на соответствующие гидроксикислоты. Натриевые соли этих кислот в слабощелочных растворах легко гидролизуются до натриевых солей соответствующих гидроксикислот. Обработка галогенокислот аммиаком дает аминокислоты.
Гидроксикислоты (оксокислоты). a-Гидроксикислоты можно получить гидролизом галогенокислот или из альдегидов (кетонов), используя синтез Килиани:

Гликолевая кислота CH2OH-COOH и молочная кислота CH3-CHOH-COOH встречаются в природе. Последняя образуется в результате молочнокислого брожения (при скисании молока) и обнаружена в тканях животных, растений и в микроорганизмах как конечный продукт окисления сахаров. b-Гидрооксикислоты можно получить кислотно-катализируемым присоединением воды к a,b-ненасыщенным кислотам. Их эфиры можно приготовить из альдегидов и кетонов по реакции Реформатского:

b-Гидроксимасляную кислоту иногда находят в виде левовращающей оптически активной формы в моче, особенно у больных сахарным диабетом. b-Гидроксикислоты довольно неустойчивы, особенно к действию сильных неорганических кислот; они легко теряют воду, образуя a,b-ненасыщенные кислоты. g-Гидроксикислоты редко встречаются в свободном состоянии; они обычно самопроизвольно переходят в устойчивые g-лактоны, которые можно получить также кислотно-катализируемой изомеризацией b,g-ненасыщенных кислот:
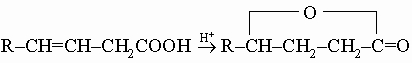
Эфиры g-гидроксикислот можно приготовить восстановлением соответствующих эфиров кетокислот.
Аминокислоты. Наиболее важными членами этой группы являются a-аминокислоты:

Многие из них можно получить гидролизом белков, в которых они, как оказалось, соединены в длинные пептидные цепи типа
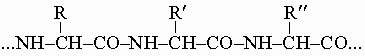
Все природные аминокислоты (кроме глицина H2NCH2COOH) оптически активны, и им приписана конфигурация
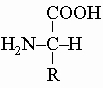
обозначаемая как L (levo). Группа R в природных аминокислотах может быть алифатической (например, в аланине R = -CH3, в серине R = -CH2OH, в цистеине R = -CH2SH, в лейцине R = -CH2CH(CH3)2), ароматической (как в фенилаланине R = -CH2C6H5 и тирозине R = -CH2C6H4OH) или гетероциклической, как в гистидине

Аминокислоты - кристаллические вещества, сильно различающиеся по растворимости в воде. Хотя все они обладают L-конфигурацией, некоторые из них являются правовращающими (+), а некоторые - левовращающими (-). Из гидролизатов белков выделено 20 аминокислот. Качественный и количественный аминокислотный состав сильно различается для разных белков. Аминокислоты можно получить действием аммиака на a-галогенокислоты или при помощи синтеза Штреккера (модификации синтеза Килиани), при котором альдегид обрабатывают синильной кислотой в присутствии хлорида аммония. Образующиеся a-аминонитрилы RCHNH2CN можно затем гидролизовать до желаемых кислот. Такие синтетические аминокислоты оптически неактивны и состоят из равных количеств право- и левовращающих форм. Разделения на оптические антиподы часто можно достичь фракционной кристаллизацией их солей с такими оптически активными основаниями, как бруцин. Аминокислоты амфотерны; в реакциях они проявляют себя то как слабые основания

,
то как слабые кислоты
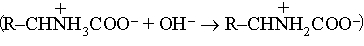
.
Восстановление эфиров аминокислот дает a-аминоспирты. При высоких температурах многие аминокислоты теряют диоксид углерода, образуя амины. Действие азотистой кислоты ведет к образованию соответствующих a-гидроксикислот, а действие нитрозилгалогенидов - к образованию a-галогенокислот. Многие из этих реакций осуществляются в более мягких условиях под действием ферментных систем живых организмов. Так, Bacillus proteus, B. coli и B. subtilis декарбоксилируют многие природные аминокислоты до аминов. Такие амины (например, путресцин H2N-(CH2)4-NH2) являются характерными продуктами разложения белков. Эти бактерии способны также заменять аминогруппу на гидроксил в мягких щелочных условиях.
5. Кетокислоты (оксокислоты, альдегидокислоты).
a-Кетокислоты. Простейшая альдегидокислота - глиоксалевая кислота HC(=O)-COOH - может быть получена гидролизом дихлоруксусной кислоты или электролитическим восстановлением щавелевой кислоты. Она встречается в природе в незрелых плодах. Простейшая a-кетокислота - пировиноградная кислота CH3COCOOH - является важным промежуточным веществом в окислительном и ферментативном расщеплении сахаров (в гликолизе) живыми организмами. Ее можно получить, используя общие реакции, ведущие к образованию a-кетокислот:

a-Кетокислоты легко окисляются:

Они также очень чувствительны к действию концентрированной серной кислоты, вызывающей реакцию

b-Кетокислоты. Свободные b-кетокислоты при нагревании легко выделяют углекислый газ, образуя кетоны:

Однако их эфиры вполне устойчивы. Их можно получить из сложных эфиров конденсацией Клайзена:

Их простейший представитель - ацетоуксусный эфир CH3COCH2COOC2H5 - имеет большое значение для органического синтеза. Его натриевая соль CH3-CO-CHNa-COOC2H5, получаемая действием этилата натрия, легко реагирует с алкилгалогенидами, давая алкилацетоуксусные эфиры, CH3COCHRCOOC2H5. Их в свою очередь можно проалкилировать до диалкилпроизводных CH3COCRR'COOC2H5. Расщепление таких эфиров до замещенных уксусных кислот можно осуществить действием сильных щелочей:
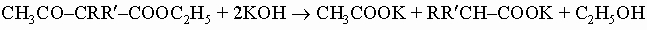
Расщепление до замещенных ацетонов достигается обработкой разбавленными кислотами или щелочами:

С реагентами, специфическими для карбонильной группы (см. разд. IV-1.А.4), b-кетоэфиры часто дают гетероциклы (см. разд. IV-4). Так, действие фенилгидразина C6H5NHNH2 ведет к пиразолонам, а гидроксиламина - к изоксазолонам.
6. Дикарбоновые кислоты принадлежат к гомологическому ряду HOOC(CH2)nCOOH (n = 0, 1, 2, 3, ...), большая часть членов которого встречается в природе. Щавелевая кислота HOOC-COOH широко распространена в растениях в форме ее солей (натрия, калия, кальция и магния). Натриевую соль получают в промышленности действием углекислого газа на натрий при 360° С. Это довольно сильная кислота (K1 = 0,04). Серная кислота превращает ее в углекислый газ, монооксид углерода и воду. Действием перманганата калия в горячем кислом растворе она количественно превращается в CO2. С ионами переходных элементов оксалат-ион образует многочисленные устойчивые комплексные ионы, например [[Fe(C2O4)3]]3-; [[Co(C2O4)3]]3-; [[Cr(C2O4)3]]3-. Последние два иона получены в оптически активной форме, что можно объяснить, только если связи металл-кислород являются по сути ковалентными и направлены к вершинам правильного октаэдра. Малоновая кислота HOOC-CH2-COOH найдена в свекольном соке. Ее можно получить из хлоруксусной кислоты, первоначально превратив последнюю в цианоуксусную кислоту NC-CH2-COOH действием NaCN с последующим гидролизом. Свободная малоновая кислота и ее C-алкилпроизводные при нагревании выше 140° С теряют CO2, давая монокарбоновые кислоты. Ее эфиры, как и ацетоуксусный эфир, легко алкилируются путем превращения в натриевую соль с последующей обработкой алкилгалогенидом:

Малоновая кислота, таким образом, очень полезна для синтеза кислот. Действуя на натриевые соли ее эфиров дигалогенидами (или галогеном), можно получить высшие дикарбоновые кислоты:

Активные атомы водорода метиленовой группы сходным образом участвуют в реакциях с карбонильными соединениями. Так, с альдегидами в присутствии пиридина как катализатора получаются a,b-ненасыщенные кислоты (реакция Кневенагеля):

Если вместо пиридина используется триэтаноламин, продуктами реакции являются b,g-ненасыщенные кислоты (модификация Линстеда). Янтарная кислота HOOC-CH2-CH2-COOH широко распространена в природе (свекольный сок, янтарь, виноград и т.д.) и образуется во многих процессах брожения. Ее получают главным образом сбраживанием тартрата аммония и малата кальция (см. ниже обсуждение яблочной кислоты под заголовками "Ненасыщенные дикарбоновые кислоты" и "Гидрокси- и кетодикарбоновые кислоты"). Ее можно получить и синтетически гидролизом ее динитрила NCCH2CH2CN, который в свою очередь получают действием цианида калия на дибромэтан. Ее ангидрид , который образуется при нагревании кислоты с ацетилхлоридом, часто используется как этерифицирующий агент, особенно если желательно отделить спирт от других веществ. Полученные таким образом полуэфиры янтарной кислоты ROOCCH2CH2COOH благодаря их кислотным свойствам могут быть легко отделены от нейтральных примесей. Регенерация спирта осуществляется омылением. Глутаровая кислота HOOC(CH2)3COOH содержится в свекольном соке и смывах с неочищенной шерсти. Ее можно получить из малонового эфира общими реакциями, приведенными выше. Адипиновая кислота HOOC(CH2)4COOH получается окислением касторового масла. Ее можно легко приготовить окислением циклогексанола (см. разд. IV-2.А.2) азотной кислотой в присутствии ванадиевого катализатора. При пиролизе этой кислоты и ее С-алкильных производных, особенно в присутствии оксидов кальция, бария и тория, образуются циклопентаноны (см. разд. IV-2.А.1). Она используется для получения найлона.
Ненасыщенные дикарбоновые кислоты. Малеиновая и фумаровая кислоты представляют собой соответственно цис- и транс-формы дегидроянтарной кислоты HOOCCH=CHCOOH и получаются при нагревании яблочной кислоты (гидроксиянтарная кислота, см. ниже).

Фумаровая кислота является главным продуктом при более низких температурах, малеиновая - при более высоких. Из этих двух кислот только малеиновая образует ангидрид, что свидетельствует о ее цис-конфигурации, тогда как в фумаровой кислоте карбоксильные группы слишком удалены друг от друга. Фумаровая кислота встречается в природе в дымянке (Fumaria officinalis) и других растениях. При термическом разложении лимонной кислоты (см. разд. IV-1.Б.7) образуются три изомерные кислоты C5H6O4:
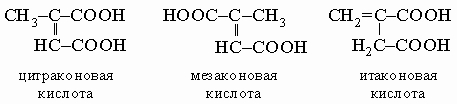
Гидрокси- (окси-) и кетодикарбоновые кислоты. Тартроновая (оксималоновая) кислота HOOC-CHOH-COOH получается в виде эфира при действии оксида серебра на броммалоновый эфир. Первым продуктом ее окисления является мезоксалевая кислота HOOC-CO-COOH. Яблочная (оксиянтарная) кислота HOOC-CHOH-CH2-COOH встречается в природе в виде оптически активной левовращающей формы во многих плодах. Мягкое окисление превращает ее в щавелевоуксусную кислоту HOOC-CO-CH2-COOH, существующую в виде цис,транс-изомерных форм. Винная кислота HOOC-CHOH-CHOH-COOH известна в четырех формах: две из них являются оптически активными зеркальными (+)- и (-)-изомерами (т. пл. 170° С); рацемическая (±)-винная (виноградная) кислота (т. пл. 204° С) представляет собой эквимолярный молекулярный комплекс (+)- и (-)-форм; мезовинная кислота (т. пл. 140° С) обладает плоскостью симметрии и потому оптически неактивна. (+)-Винная кислота получается из вина в виде ее кислой калиевой соли. При обработке щелочью она превращается в смесь (+)-, (-)- и мезо-форм.
7. Трикарбоновые кислоты. Трикарбаллиловая кислота HOOC-CH2-CH(COOH)-CH2-COOH получается гидролизом ее тринитрила NC-CH2-CH(CN)-CH2-CN, образующегося при действии цианида калия на 1,2,3-трибромпропан. Она также является продуктом гидрирования аконитовой кислоты, возникающей при дегидратации лимонной кислоты. Лимонная кислота HOOC-CH2-C(OH)(COOH)-CH2-COOH найдена во многих фруктовых соках. Ее получают в промышленных масштабах действием микроорганизмов (цитромицетов) на такие углеводы, как мальтоза. Она находит многочисленные применения в приготовлении искусственных фруктовых напитков. Ее дегидратация дает аконитовую кислоту HOOC-CH=C(COOH)-CH2-COOH.
Энциклопедия Кольера. — Открытое общество. 2000.
Полезное
Смотреть что такое "АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ" в других словарях:
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, в органической химии термин, используемый для описания любого соединения, у которого в структуре не содержится никаких колец. см. также ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ … Научно-технический энциклопедический словарь
Ациклические соединения — (соединения жирного ряда или алифатические) органические соединения, в молекулах которых отсутствуют циклы и все атомы углерода соединены между собой в прямые или разветвленные (открытые) цепи. Различают две основные группы ациклических… … Википедия
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — то же, что алифатические соединения … Большой Энциклопедический словарь
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, см. Алифатические соединения … Большая медицинская энциклопедия
ациклические соединения — то же, что алифатические соединения. * * * АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, то же, что алифатические соединения (см. АЛИФАТИЧЕСКИЕ СОЕДИНЕНИЯ) … Энциклопедический словарь
ациклические соединения — (см. а... + циклический) то же, что алифатические соединения. Новый словарь иностранных слов. by EdwART, , 2009. ациклические соединения [гр. частица отрицания + циклический] – хим. (жирные или алифатические) – обширный класс органических… … Словарь иностранных слов русского языка
Ациклические соединения — соединения жирного ряда, или алифатические соединения, органические вещества (углеводороды и их производные), молекулы которых не содержат циклов, а представляют собой «открытые» цепи. В соединениях т. н. нормального строения,… … Большая советская энциклопедия
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — (от греч. а приставка, означающая отрицание, и kyklos круг, цикл), алифатические соединения, жирного ряда соединения, органич. соединения, в молекуле к рых атомы углерода связаны между собой в открытые разветвлённые или неразветвлённые цепи (см … Большой энциклопедический политехнический словарь
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — то же, что алифатические соединения … Химическая энциклопедия
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — то же, что алифатические соединения … Естествознание. Энциклопедический словарь
 18+© Академик, 2000-2025
18+© Академик, 2000-2025- Обратная связь: Техподдержка, Реклама на сайте
Экспорт словарей на сайты, сделанные на PHP, Joomla, Drupal, WordPress, MODx.