- КВАНТОВАЯ ХИМИЯ
- КВАНТОВАЯ ХИМИЯ
-
область теор. химии, в к-рой идеи и методы квант. механики применяются к исследованию атомов, молекул и др. хим. объектов и процессов. Квантовомеханич. подход в химии чаще всего основывается на Шредингера уравнении для атома, молекулы или совокупности атомов и молекул: Hy=Ey. Оператор Н (гамильтониан) учитывает как кинетич. энергию составляющих систему ч-ц (ат. ядер и эл-нов), так и энергию их вз-ствия друг с другом и с внеш. полями. Решение ур-ння даёт значение полной энергии системы Е и её состояния — волновые ф-ции y, к-рые зависят от пространств. и спиновых координат всех ч-ц и с помощью к-рых можно в принципе рассчитать св-ва системы. Однако точные решения найдены лишь для атома водорода (см. КВАНТОВАЯ МЕХАНИКА), поэтому для решения конкретных задач К. х. разработан ряд приближённых методов.Электронное строение молекул — гл. предмет К. х. Согласно адиабатич. приближению, движение эл-нов в ат. системах рассматривается при фиксиров. положениях ядер и описывается электронной волн. ф-цией, зависящей от координат эл-нов и ядер. Из неполных сведений о виде этой ф-ции можно вывести качеств. интерпретацию физ. св-в молекул и их спектров, а более точные вычисления позволяют получить количеств. результаты.Основы квант. теории многоэлектронных систем были заложены в работе нем. физика В. Гейзенберга, посвященной атому гелия (1926), и работах нем. физиков В. Гейтлера (Хайтлер) и Ф. Лондона о молекуле водорода (1927). Они показали, что существование, устойчивость и св-ва этих систем невозможно объяснить в рамках классич. представлений. В последующих исследованиях были развиты методы определения электронных волн. ф-ций для более сложных ат. систем. Наиболее важный из них — метод мол. орбиталей (МО) — рассматривает движение валентных эл-нов молекулы в ноле всех остальных эл-нов и ядер атомов, входящих в молекулу. Волн. ф-ции при таком одноэлектронном приближении находят при решении ур-ния Шрёдингера вариац. методом, обычно по схеме самосогласованного поля.. Метод МО представляет собой упрощённый вариант более общего метода вз-ствия конфигураций, к-рый в принципе позволяет рассчитывать достаточно точные волновые ф-ции молекул.Нахождение и использование даже простейших волновых ф-ций сопряжено с весьма трудоёмкими вычислениями.В ранних квантовохим. исследованиях применялись почти исключительно приближённые полуэмпирич. методы. В сочетании с возмущений теорией они развивались как искусство делать качеств. предсказания практически без вычислений, основываясь на интуиции и аналогиях. Так были установлены принципы теории межатомных взаимодействий и межмолекулярных взаимодействий, разработаны основы мол. спектроскопии, создана качеств. теория строения и реакц. способности нек-рых типов органич. молекул.Развитие вычислительной техники в 60-х гг. 20 в. изменило стиль и направление квантовохим. исследований. Стали быстро развиваться неэмпирич. методы расчёта молекул и количеств. варианты полуэмпирич. методов. Расчёт на ЭВМ электронного строения молекул ср. размеров (20—30 эл-нов) производится уже с точностью, во мн. случаях достаточной для предсказания геом. строения, физ. св-в и спектров таких молекул. Особенно важны квантовохим. методы расчёта при изучении не поддающихся эксперим. регистрации короткоживущих активных ч-ц и активированных комплексов.На совр. этапе в К. х. наряду с традиц. расчётами электронных волн. ф-ций разрабатываются новые проблемы и методы. Развивается квант. теория движения ядер в хим. системах, рассматриваются системы, меняющиеся во времени — в условиях хим. реакций, фотовозбуждения и распада и т. д. Успешное решение задач К. х. во многом зависит от развития методов квант. механики и статистич. физики так, что К. х. можно с основанием рассматривать как ветвь теор. физики.
Физический энциклопедический словарь. — М.: Советская энциклопедия. Главный редактор А. М. Прохоров. 1983.
- КВАНТОВАЯ ХИМИЯ
-
- область теоретич. химии, изучающая строение и хим. превращение атомов, молекул и др. многоатомных систем на основе квантовой механики. Осн. ур-ние К. х.- нерелятивистское Шрёдингера уравнение:
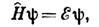
где y - волновая ф-ция системы, зависящая от пространств. и спиновых координат всех частиц системы. |y|2 характеризует пространств. распределение электронов и ядер в ней, E - полная внутренняя энергиясистемы. Гамильтониан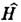 для молекулы имеет вид
для молекулы имеет вид 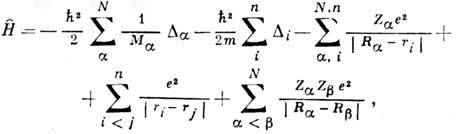
где первый член описывает кинетич. энергию ядер, второй - кинетич. энергию электронов, третий - энергию их эл.-статич. притяжения ядрами, четвёртый - энергию взаимодействия электронов между собой, пятый-межъядерное отталкивание, Da и Di - операторы Лапласа, Мa - масса ядра атома a, т - масса электрона, е - его заряд, Za и Zb - зарядовые числа ядер атомов a и b, Ra и Rb - координаты этих ядер, ri и rj - координаты i -го и j -гoэлектронов, п - число электронов, N- число атомов в молекуле. Решения ур-ния Шрёдингера дают значения полной энергии системы Eи волновой ф-ции y. Однако точные аналитич. решения получены только для атома водорода. Для более сложных систем при решении ур-ния Шрёдингера используют ряд последоват. приближений и численное решение на ЭВМ полученных ур-ний. <В первом - адиабатич.- приближении, предложенном М. Борном (М. Born) и Р. Оппенгеймером (R. Oppenheimer) в 1927, полагают, что движение электронов можно рассматривать как независимое от медленного движения ядер, т. к. массы ядер значительно (на 3-4 порядка) превышают массу электронов. Решение задачи в этом случае разбивается на два этапа: сначала решают ур-ние Шрёдингера только для электронной части гамильтониана при фиксированном положении ядер. При этом волновая ф-ция должна быть антисимметричной по отношению к перестановке электронов, т. е. при перестановке двух электронов с одинаковыми спинами полная волновая ф-ция должна менять знак (см. Паули принцип). Суммарная энергия взаимодействия ядер с электронами, электронов между собой и взаимодействия неподвижных атомных ядер является потенц. энергией ядер. Зависимость потенц. энергии ядер от их координат образует потенциальную поверхность(3N-5)-мерную для линейных и (3N-6)-мерную для всех остальных молекул, состоящих из N атомов. На этом этапе получают энергии основного и возбуждённых электронных состояний молекул. Затем решают задачу о движении (колебании) ядер в поле потенциала, полученного при решении предыдущей задачи, при этом получают значения колебат. энергии молекулы. <Основы квантовой теории многоэлектронных систем были заложены в работе В. Гейзенберга (W. Heisenberg; 1926), посвящённой атому гелия, а также в работах В. Гайтлера (W. Heitler) и Ф. Лондона (F. London) о молекуле водорода (1927). Они показали, что существование, устойчивость и свойства этих систем невозможно объяснить в рамках классич. представлений. Согласно В. Гайтлеру и Ф. Лондону, связывание между атомами в молекуле водорода обусловлено т. н. обменным взаимодействием. Дальнейшее развитие теории многоэлектронных атомов связано с методом самосогласованного поля, предложенного в 1927 Д. Р. Хартри (D. R. Hartree). В нём взаимодействие каждого из электронов со всеми остальными заменяется взаимодействием с усреднённым полем, создаваемым остальными электронами. В 1930 В. А. Фок усовершенствовал метод Хартри, использовав для многоэлектронной волновой ф-ции представление в виде слейтеровского детерминанта: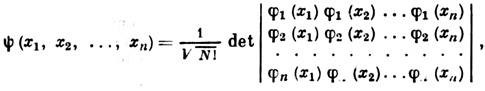
где j j (xj) - одноэлектронная спин-орбиталь (см. Молекулярная орбиталь), xj-(rj,aj), где rj - пространств. координаты, aj - спиновые координаты электрона. Такой вид волновой ф-ции позволяет учесть принцип Паули. Одноэлектронные ф-ции (орбитали) находят, решая ур-ния Хартри - Фока (см. Хартри - Фока метод):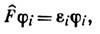 где
где 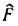 - оператор, наз. фокианом, ei - энергии i -йзаполненной орбитали (рассматриваются состояния системы, полный спин к-рой равен нулю). Энергия системы в этом случае равна:
- оператор, наз. фокианом, ei - энергии i -йзаполненной орбитали (рассматриваются состояния системы, полный спин к-рой равен нулю). Энергия системы в этом случае равна: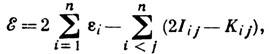
где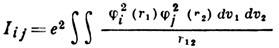
и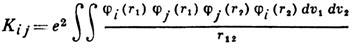
- соответственно кулоновский и обменный интегралы, представляющие собой ср. энергию эл.-статич. отталкивания и обменного взаимодействия пары электронов, находящихся на i -й и j -й орбиталях, п - число электронных пар, v1 и v2 - пространств. объёмы, в к-рых изменяются координаты первого и второго электронов соответственно. Система ур-ний Хартри - Фока является системой нелинейных интегродифференц. ур-ний. Нелинейность ур-ний означает, что их решения ji есть собств. ф-ции оператора
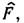 к-рый, в свою очередь, определяется через орбитали ji. Эта особенность ур-ний Хартри - Фока позволяет решать их итераций методом. В 1927-29 Ф. Хунд (F. Hund) и Р. С. Малликен (R. S. Mulliken) развили идею нового подхода к поиску волновой ф-ции молекулы - т. н. метод молекулярных орбиталей (МО). Метод МО рассматривает движение электронов молекулы в поле, создаваемом всеми остальными электронами и ядрами атомов молекулы. Полная энергия молекулы с волновой ф-цией в виде МО определяется соотношением
к-рый, в свою очередь, определяется через орбитали ji. Эта особенность ур-ний Хартри - Фока позволяет решать их итераций методом. В 1927-29 Ф. Хунд (F. Hund) и Р. С. Малликен (R. S. Mulliken) развили идею нового подхода к поиску волновой ф-ции молекулы - т. н. метод молекулярных орбиталей (МО). Метод МО рассматривает движение электронов молекулы в поле, создаваемом всеми остальными электронами и ядрами атомов молекулы. Полная энергия молекулы с волновой ф-цией в виде МО определяется соотношением 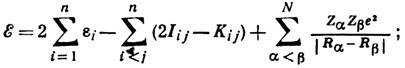
энергия МО ei является энергией электрона, находящегося на i -й МО. Для нахождения одноэлектронной ф-ции МО можно использовать метод Хартри - Фока, однако практич. решение сложно и проводится только для атомов и двухатомных молекул. Для всех остальных систем используют приближение, предложенное С. С. Рутаном (С. С. Roothaan; 1951): атомные орбитали обычно представляют в виде разложения по базисным ф-циям cm слейтеровского или гауссовского типа, также центрированным на ядрах: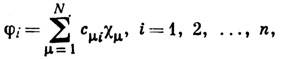
и вместо самих ф-ций ji оптимизирует коэффициенты сmi . В результате система интегродифференц. ур-ний Хартри - Фока переходит в систему алгебраич. ур-ний Хартри - Фока - Рутана. Эти ур-ния положены в основу алгоритмов всех неэмпирических программ К. х. <Один из важных результатов теории Хартри - Фока - теорема Купменса: энергия орбитали ei, получаемая при решении ур-ний Хартри - Фока, даётприближённое значение потенциала ионизации электрона, занимающего i -ю МО. В эксперим. исследованиях наблюдается последовательность (по энергиям) ионизованных состояний. Теорема Купменса позволяет приближённо интерпретировать эту последовательность как отрыв электронов с последовательных МО. Теорему Купменса используют при интерпретации эксперим. данных фотоэлектронной и рентгеноэлектронной спектроскопии. <Процесс возбуждения электрона можно рассматривать и как переход электрона с заполненной МО на вакантную. В этом случае для оценки энергии возбуждения DE возб можно использовать соотношение DE возб~-ei+e а, где ei - энергия заполненной МО, а e а - энергия вакантной МО. Такой подход используется при интерпретации спектров электронного возбуждения, оже-спектров и т. д. <Качеств. представления о структуре МО (т. н. узловой структуре) лежат в основе мн. теорий формы молекул и протекания хим. реакций. Наиб. известной теорией качеств. формы молекул в приближении МО является теория Уолша, в основу к-рой положено соотношение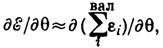 где q - валентный уголмолекулы. Для предсказания формы молекул необходимо знать, как зависит энергия МО от внутр. координат: если сумма энергий МО при изгибании линейной молекулы будет понижаться, то её устойчивая конфигурация угловая, причём угол q может быть рассчитан (рис.).
где q - валентный уголмолекулы. Для предсказания формы молекул необходимо знать, как зависит энергия МО от внутр. координат: если сумма энергий МО при изгибании линейной молекулы будет понижаться, то её устойчивая конфигурация угловая, причём угол q может быть рассчитан (рис.).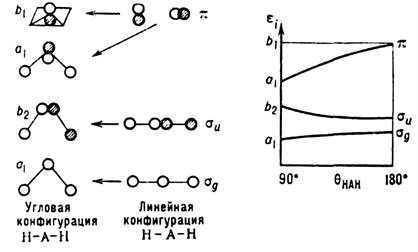
Протекание хим. реакций качественно объясняют на основе метода МО в рамках правил Вудворда - Гоффмана, правил Пирсона, метода Фукуи и др.; наиболее часто используют правила Вудворда - Гоффмана. Применяя эти правила, строят корреляц. диаграммы хим. реакций, для этого: выделяют хим. связи и орбитали, к-рые наиболее сильно изменяются в ходе реакции; задав путь сближения реагентов или отд. связей при внутримолекулярных реакциях в направлении формирования ожидаемой структуры переходного состояния, классифицируют МО реагентов и продуктов по свойствам симметрии, присущей выбранной структуре переходного состояния; устанавливают соответствие между МО реагентов и продуктов в рамках выбранной классификации орбиталей по симметрии. При этом учитывают оси и плоскости симметрии, проходящие через рвущиеся и образующиеся в ходе реакции связи. Если в процессе хим. реакции все заполненные МО реагентов переходят в заполненные МО продуктов в осн. состоянии (сохраняется орбитальная симметрия), то данная реакция наз. разрешённой. Если в процессе такой реакции происходит пересечение уровней заполненных и вакантных МО реагентов и продуктов, то реакция наз. запрещённой. <Рассмотрение протекания хим. реакций на основе правил Вудворда - Гоффмана носит качеств. характер, запрет по симметрии не означает невозможности протекания реакции в данном электронном состоянии. Однако запрещённые по симметрии реакции имеютвысокие потенц. барьеры или включают промежуточное образование радикалов (или ионов). Реакции же, разрешённые по симметрии в осн. состоянии, имеют, как правило, низкие потенц. барьеры или не имеют их совсем. <Последоват. решение ур-ний Хартри - Фока - Рутана на ЭВМ лежит в основе неэмпирич. методов К. х. Однако вычисление волновых ф-ций и энергий в приближении Хартри - Фока - Рутана сопряжено со значит. трудностями, т. к. число интегралов, описывающих межэлектронное отталкивание, при увеличении размеров молекулярной системы растёт как N4, где N - число базисных ф-ций. Поэтому для сложных систем применяют полуэмпирич. методы, в к-рых большая часть интегралов заменяется экспериментально полученными данными (потенциалы ионизации и сродство к электрону атома). Эти методы были распространены в ранних квантовомеханич. исследованиях. <Приближение Хартри - Фока - Рутана во мн. случаях даёт большие погрешности (напр., отрицат. значение энергии связи для F2, неправильную симметрию для осн. электронного состояния молекулы С 2, неправильный знак для дипольного момента СО; приводит к неправильной последовательности ионизированных состояний молекул F2, N2 и т. д.). Для устранения недостатков этого метода учитывают энергии корреляции электронов, что позволяет определить отклонение идеализированной одноэлектронной модели от реальной. <Для учёта энергии корреляции электронов в неэмпирич. расчётах чаще всего используют два подхода: метод конфигурац. взаимодействия и теорию возмущений. В методе конфигурац. взаимодействия волновая ф-ция записывается в виде линейной комбинации слейтеровских детерминантов yk, отвечающих разным заполнениям МО:
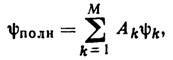
где М - число учитываемых конфигураций. Волновые ф-ции при таком подходе находят при решении электронной части ур-ния Шрёдингера вариац. методом. В теории возмущений точные решения ур-ния Шрёдингера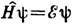 ищутся на основе известных решений ур-ния Шрёдингера H(0)y(0)=E(0)y(0) с модельным гамильтонианом
ищутся на основе известных решений ур-ния Шрёдингера H(0)y(0)=E(0)y(0) с модельным гамильтонианом 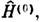 к-рый отличается от точного на малое возмущение w; волновую ф-цию и энергию ищут в виде рядов:
к-рый отличается от точного на малое возмущение w; волновую ф-цию и энергию ищут в виде рядов:y =y(0)+y(1) + y(2)+...
E = E(0) +E(1)+ E(2)+...В квантовохим. расчётах за
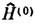 выбирают гамильтониан Хартри - Фока - Рутана, а за возмущение w- оператор, описывающий электронную корреляцию. Оба эти метода позволяют, в принципе, получать практически точные решения нерелятивистского ур-ния Шрёдингера. <Прогресс в вычислит. технике стимулировал развитие неэмпирич. методов К. х., в т. ч. методов, учитывающих энергию электронной корреляции. Высокая точность неэмпирич. расчётов мн. молекулярных характеристик позволила изучать свойства молекул независимо от эксперимента, что привело к ряду принципиально новых результатов. Так, одним из неопровержимых постулатов считалось представление о том, что макс. валентность атома углерода равна 4 и, следовательно, в соединениях с одновалентными лигандами его координац. число равно 4. Однако с помощью неэмпирич. расчётов П. Р. Шлейер (P. R. Schleyer; 1982- 83) предсказал существование стабильных молекул СLi5 и CLi6, к-рые позднее были экспериментально обнаружены. С помощью расчётов было открыто большое число структурно-нежёстких молекул, в к-рых обнаруженыколебания ядер большой амплитуды (порядка длины хим. связи) для валентносвязанных атомов. В таких молекулах нарушается традиц. деление взаимодействий на валентные и невалентные, и их невозможно описать в рамках классич. представлений. На основе неэмпирич. расчётов было также идентифицировано большое число молекул в межзвёздном пространстве. На теоретически рассчитанных значениях атом-атомных потенциалов основаны современные исследования структуры и термодинамики жидкостей, расплавов и растворов. <В рамках К. х., наряду с традиц. расчётами геом. и электронной структуры молекул, развиваются квантовая теория полимерных молекул, движения ядер в ходе хим. реакции, теория фотовозбуждения и т. п. Успешное развитие методов К. х. во многом зависит от развития методов квантовой механики, квантовой теории поля и статистич. физики, методов вычислит. математики. Лит.: Фок В. А., Начала квантовой механики, 2 изд., М., 1976; С л э т е р Д ж., Методы самосогласованного поля для молекул и твердых тел, пер. с англ., М., 1978; Минкин В. И., Симкин Б. Я., Миняев Р. М., Теория строения молекул, М., 1979; Ф у д з и н а г а С., Метод молекулярных орбиталей, пер. с япон., М., 1983. А. И. Болдырев, А. А. Овчинников.
выбирают гамильтониан Хартри - Фока - Рутана, а за возмущение w- оператор, описывающий электронную корреляцию. Оба эти метода позволяют, в принципе, получать практически точные решения нерелятивистского ур-ния Шрёдингера. <Прогресс в вычислит. технике стимулировал развитие неэмпирич. методов К. х., в т. ч. методов, учитывающих энергию электронной корреляции. Высокая точность неэмпирич. расчётов мн. молекулярных характеристик позволила изучать свойства молекул независимо от эксперимента, что привело к ряду принципиально новых результатов. Так, одним из неопровержимых постулатов считалось представление о том, что макс. валентность атома углерода равна 4 и, следовательно, в соединениях с одновалентными лигандами его координац. число равно 4. Однако с помощью неэмпирич. расчётов П. Р. Шлейер (P. R. Schleyer; 1982- 83) предсказал существование стабильных молекул СLi5 и CLi6, к-рые позднее были экспериментально обнаружены. С помощью расчётов было открыто большое число структурно-нежёстких молекул, в к-рых обнаруженыколебания ядер большой амплитуды (порядка длины хим. связи) для валентносвязанных атомов. В таких молекулах нарушается традиц. деление взаимодействий на валентные и невалентные, и их невозможно описать в рамках классич. представлений. На основе неэмпирич. расчётов было также идентифицировано большое число молекул в межзвёздном пространстве. На теоретически рассчитанных значениях атом-атомных потенциалов основаны современные исследования структуры и термодинамики жидкостей, расплавов и растворов. <В рамках К. х., наряду с традиц. расчётами геом. и электронной структуры молекул, развиваются квантовая теория полимерных молекул, движения ядер в ходе хим. реакции, теория фотовозбуждения и т. п. Успешное развитие методов К. х. во многом зависит от развития методов квантовой механики, квантовой теории поля и статистич. физики, методов вычислит. математики. Лит.: Фок В. А., Начала квантовой механики, 2 изд., М., 1976; С л э т е р Д ж., Методы самосогласованного поля для молекул и твердых тел, пер. с англ., М., 1978; Минкин В. И., Симкин Б. Я., Миняев Р. М., Теория строения молекул, М., 1979; Ф у д з и н а г а С., Метод молекулярных орбиталей, пер. с япон., М., 1983. А. И. Болдырев, А. А. Овчинников.
Физическая энциклопедия. В 5-ти томах. — М.: Советская энциклопедия. Главный редактор А. М. Прохоров. 1988.
.