- Марфана синдром
-
I
Марфа́на синдро́м (В. J.A. Marfan, франц. педиатр, 1858—1942; синоним болезнь Марфана)
наследственное системное поражение соединительной ткани, проявляющееся «патологическими изменениями опорно-двигательного аппарата, глаз и сердечно-сосудистой системы. Впервые синдром описал Марфан в 1896 г. Встречается редко.Заболевание наследуется по аутосомно-доминантному типу. Патогенетические механизмы окончательно не выяснены. Установлено, что при М.с. основной дефект связан с нарушениями коллагена, хотя не исключена возможность поражения эластических волокон соединительной ткани. Оба пола поражаются одинаково часто.Отдельные клинические признаки М.с. могут наблюдаться уже при рождении, например арахнодактилия — удлинение пальцев кистей и стоп (рис. 1), но наиболее ярко симптомокомплекс проявляется у детей школьного возраста (рис. 2). У больных резко выражен астенический тип сложения (высокий рост, истончение подкожной клетчатки, мышечная слабость). Характерными признаками М.с. являются долихоцефалия — изменение формы головы, когда продольный размер значительно превышает поперечный, так называемое птичье лицо — узкое, с близкорасположенными глазами, тонким носом и выступающей вперед верхней челюстью (прогнатия); деформация ушных раковин, высокое небо. Иногда наблюдается расщепление твердого неба (волчья пасть). Конечности, пальцы кистей и стоп удлинены, грудная клетка воронкообразной или килевидной формы, ребра тонкие и длинные, межреберные промежутки широкие, позвоночник искривлен (Сколиоз, Кифоз или Лордоз), отмечаются разболтанность суставов, иногда с переразгибанием в коленных суставах, плоскостопие. При рентгенологическом исследовании костей выявляют истончение коркового слоя и костных перекладин.Из поражений глаз наиболее часто встречается вывих или подвывих хрусталика, обусловливающий высокую степень миопии (см. Близорукость) или гиперметропии (см. Дальнозоркость). Нередко обнаруживают отсутствие ресниц, голубую окраску склер, Косоглазие, гетерохромию (неодинаковую окраску) радужки, катаракту (Катаракта), дистрофические изменения сетчатки глаза. У больных с М.с. отмечаются признаки вегетативно-сосудистой дистонии (Вегетативно-сосудистая дистония): потливость, похолодание конечностей, мраморный рисунок кожи, акроцианоз. При исследовании сердца часто диагностируют изменения в проводящей системе, метаболические нарушения в миокарде, расширение просвета аорты и др. Нередко формируется Аневризма аорты или легочного ствола. В некоторых случаях наблюдаются пороки развития легких (например. уменьшение числа долей), эндокринные расстройства: Акромегалия, несахарный диабет (см. Диабет несахарный), нарушение менструального цикла (Менструальный цикл). Интеллект у больных с М.с., как правило, сохранен.Диагноз ставят на основании клинической картины. Наиболее часто М.с. дифференцируют с врожденной контрактурной арахнодактилией. Это заболевание наследуется по аутосомно-доминантному типу и проявляется контрактурами локтевых, коленных и межфаланговых суставов, арахнодактилией, кифосколиозом; поражения глаз отсутствуют. М.с. следует отличать от доминантно наследуемой артроофтальмопатии (синдрома Стиклера). Эти два синдрома сближают такие симптомы, как астеническое телосложение, длинные тонкие пальцы, повышенная гибкость суставов. Однако при синдроме Стиклера отмечаются увеличение размеров крупных суставов и рентгенологические признаки эпифизарной дисплазии с расширением дистальных концов длинных трубчатых костей, аномалии ротолицевой области («плоское» лицо, гипоплазия средней части лица и нижней челюсти, аномальный рост зубов, расщелина неба). Аномалии глаз при синдроме Стиклера сводятся к миопии, катаракте, отслойке и дегенерации сетчатки, что может привести к слепоте; вывиха хрусталика, характерного для М.с., никогда не наблюдается.Лечение симптоматическое. Прогноз определяется в основном изменениями сердечно-сосудистой системы. Причинами смерти чаще всего являются разрыв аневризмы аорты или легочного ствола.Профилактика заключается в своевременном проведении медико-генетического консультирования (Медико-генетическое консультирование) в семьях, где имелись больные с синдромом Марфана.См. также Наследственные болезни.Библиогр.: Наследственная и приобретенная патология обмена веществ у детей, под ред. В.А. Таболина, с. 122, М., 1971, Руководство по педиатрии, под ред. Р.Е. Бермана и В.К. Вогана, пер. с англ., кн. 6, с. 371, М., 1987.
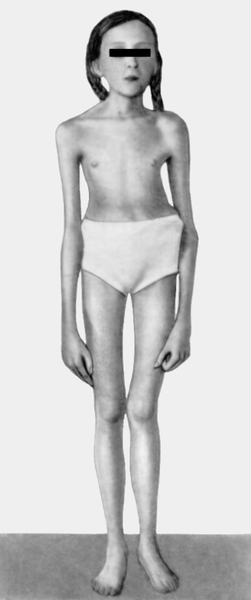 клетка, искривление позвоночника">Рис. 2. Девочка с выраженными признаками синдрома Марфана: астеническое телосложение, «птичье лицо», конечности удлинены, арахнодактилия, деформированная грудная клетка, искривление позвоночника.
клетка, искривление позвоночника">Рис. 2. Девочка с выраженными признаками синдрома Марфана: астеническое телосложение, «птичье лицо», конечности удлинены, арахнодактилия, деформированная грудная клетка, искривление позвоночника.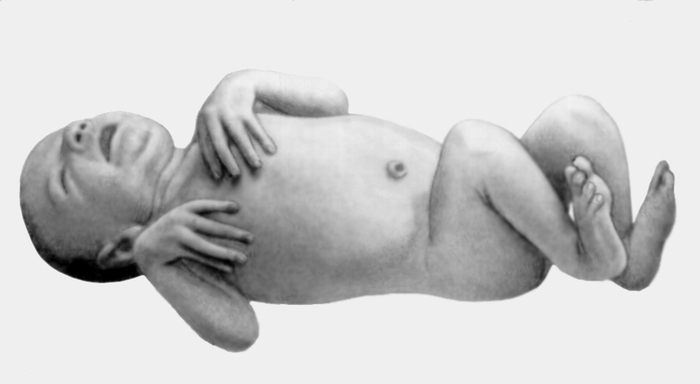 Рис. 1. Новорожденный с синдромом Марфана: арахнодактилия кистей и стоп.II Марфа́на синдро́м (В.J.A. Marfan)наследственная болезнь, обусловленная аномалией развития соединительной ткани и характеризующаяся сочетанием поражений опорно-двигательного аппарата (чрезмерно длинные конечности, арахнодактилия и др.), подвывиха или вывиха хрусталиков и вегетативно-сосудистых расстройств; наследуется по аутосомно-доминантному типу.
Рис. 1. Новорожденный с синдромом Марфана: арахнодактилия кистей и стоп.II Марфа́на синдро́м (В.J.A. Marfan)наследственная болезнь, обусловленная аномалией развития соединительной ткани и характеризующаяся сочетанием поражений опорно-двигательного аппарата (чрезмерно длинные конечности, арахнодактилия и др.), подвывиха или вывиха хрусталиков и вегетативно-сосудистых расстройств; наследуется по аутосомно-доминантному типу.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг.