- ХИМИЯ АНАЛИТИЧЕСКАЯ
наука о методах определения химического состава веществ. Химический анализ буквально пронизывает всю нашу жизнь. Его методами проводят скрупулезную проверку лекарственных препаратов. В сельском хозяйстве с его помощью определяют кислотность почв и содержание в них питательных веществ, что позволяет подобрать оптимальные условия обработки почвы, также оценивают содержание белка и влаги в разных сортах зерна. Химическому анализу подвергаются и товары широкого потребления: в зубной пасте контролируют содержание фтора, в маслах - содержание ненасыщенных соединений. В природоохранной деятельности методы аналитической химии применяют для контроля качества питьевой воды, для определения содержания вредных веществ в отходах и т.д. В судебной практике с их помощью обнаруживают следы пороха на руках подозреваемого, анализируют состав красок, которыми написана картина, чтобы отличить подлинник от подделки. Методы анализа различаются по степени сложности. Так, в медицине используются экспресс-тесты на беременность и сложные методы анализа крови на содержание сахара или холестерина, контроля уровня нейромедиаторов при исследовании мозга in vivo и пр. Из приведенных примеров видно, что все вопросы, которые решает аналитическая химия, можно свести к следующим: что представляет собой данное вещество, из каких компонентов оно состоит, каковы их количество и распределение? Чтобы ответить на эти вопросы, проводят самые разнообразные химические реакции, применяют широкий спектр химических, физических, физико-химических, биологических методов, разрабатывают новые методы анализа и совершенствуют уже существующие. Число методов аналитической химии чрезвычайно велико и постоянно растет. Аналитическая химия тесно связана с другими дисциплинами: химический анализ внедряется в различные области науки, химик-аналитик пользуется достижениями других разделов химии, а также математики, физики, биологии и многих областей техники.
ОСНОВНЫЕ ПОЛОЖЕНИЯ
Стадии анализа. Решение аналитических задач включает несколько стадий.
Постановка задачи. Эта несущественная на первый взгляд стадия на самом деле очень важна. Предположим, нужно определить количество ртути в водоеме. А что именно подразумевается под словом "ртуть"? Это может быть вся ртуть, независимо от конкретной химической формы, или все органические соединения ртути (например, диметилртуть), или все ее неорганические соединения, или вся ртуть в определенной степени окисления, или идентификация всех ртутьсодержащих соединений и определение их количества. Аналогичным образом обстоит дело и с "водоемом". Следует ли ограничить определение растворенной ртутью или рассмотреть взвешенные в воде твердые частички, ил на дне водоема, обитающих в воде животных и растения? Нужно учесть и продолжительность анализа: достаточно ли единичное определение, или потребуется рассчитать среднюю величину из результатов нескольких измерений, сделанных в течение одного дня, а может быть, и целого года. Ответы на эти вопросы определят характер всего анализа.
Выбор метода. Метод анализа выбирают исходя из поставленной задачи, размеров объекта и образца, содержания определяемых веществ, наличия примесей, требуемой точности результатов и имеющегося оборудования; учитывают также возможную продолжительность и стоимость анализа. Рассмотрим, например, два случая определения свинца. В первом - по результатам анализа устанавливают стоимость переработки руды, которая зависит от содержания свинца. Имеется большой образец, концентрация свинца в нем высокая, ответ необходим точный. Во втором случае нужно определить, загрязнен ли свинцом металл, из которого изготовлена старинная монета. Содержание свинца низкое, требуется лишь приблизительная его оценка, в ходе анализа сама монета не должна пострадать. Понятно, что эти случаи требуют разного подхода. Для анализа образца руды можно применить такие методы, как гравиметрия или титрование. Для монеты потребуется другой, щадящий (неразрушающий) метод, например флуоресценция в рентгеновских лучах.
Отбор образца. Для разных аналитических методов требуются, конечно, и разные по величине образцы - в количестве от нанограммов (1 нг = 10-9 г) до нескольких граммов. Вряд ли возможно целиком проанализировать объект, который весит намного больше, чем требует выбранная для анализа методика. В этих случаях отбирают образец, или пробу, вещества. Эта проба должна быть репрезентативной, т.е. адекватной всему объекту или той его части, которая представляет наибольший интерес. В приведенном выше примере со ртутью в водоеме постановка задачи определяет и способ отбора пробы.
Подготовка образца к анализу. Если количественные измерения проводят в растворе, образец растворяют в подходящем растворителе; при этом концентрацию образца подбирают так, чтобы она находилась в пределах применимости метода. Иногда приходится выделять определяемое вещество из смеси, поскольку многие методы анализа неспецифичны и даже неселективны. Специфичным называют метод, при помощи которого определяется только конкретное вещество, а селективным - предпочтительный для данного вещества метод, пользуясь которым можно определять и другие вещества. Специфичных методов очень мало, селективных - значительно больше. Например, высоко-селективны масс-спектрометрия и иммунологический анализ.
Измерения. Чтобы определить количество анализируемого вещества или его состав, измеряют какую-либо его физическую величину: количество вещества, израсходованного или образовавшегося в результате химической реакции; скорость реакции; интенсивность поглощения, испускания или рассеяния света; ток, возникающий в ходе окислительно-восстановительных процессов; количество выделившегося или поглощенного тепла и т.д. Зная связь между результатами измерений и теми величинами, которые интересуют исследователя, а также сравнив эти результаты с соответствующими стандартами, устанавливают количество определяемого вещества или его состав.
Интерпретация результатов. Когда результаты уже получены, может возникнуть ряд вопросов: решена ли поставленная задача? как проводить дальнейшие исследования? Не исключено, что для получения более точных результатов нужно усовершенствовать методику анализа.
Рабочие кривые. Рабочая кривая - это графическая зависимость, связывающая концентрацию определяемого вещества с тем параметром, который измеряется в ходе анализа (оптической плотностью, интенсивностью флуоресценции, электродным потенциалом, скоростью реакции и т.д.). Масштаб координатных осей - линейный или логарифмический - выбирается в зависимости от конкретного эксперимента. Логарифмические оси используют, в частности, при изменении концентрации в широких пределах. Если нужны более точные результаты, предпочтительны линейные оси и узкие интервалы концентрации. Для построения рабочей кривой сначала готовят стандартные образцы известной концентрации. Затем для каждого из них измеряют тот или иной параметр и откладывают его значение в виде точки против соответствующей концентрации. По точкам проводят плавную кривую, на которую точки ложатся наилучшим образом. Для этого используют какую-либо подходящую математическую функцию или эмпирическую зависимость. Затем измеряют тот же параметр для исследуемого образца и по рабочей кривой определяют его концентрацию (рис. 1). У каждого метода есть свои рабочий диапазон, чувствительность, фон, порог обнаружения.
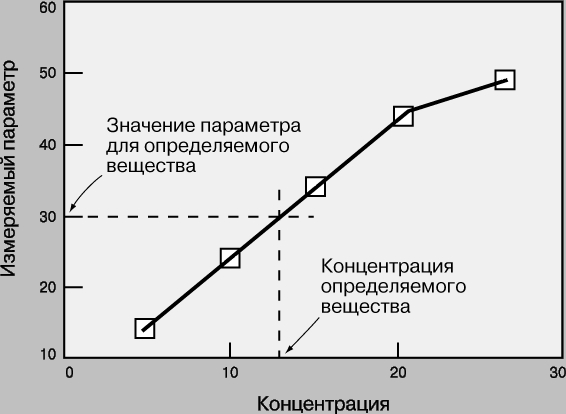
Рис. 1. РАБОЧАЯ КРИВАЯ - зависимость измеряемого параметра от концентрации для стандартного вещества. С ее помощью можно найти концентрацию определяемого вещества, соответствующую данному значению параметра.
Рабочий диапазон - это диапазон концентраций, в пределах которого применима данная методика. Линейный участок кривой отвечает области концентраций, в которой результаты наиболее надежны. При близких к предельным высоких и низких концентрациях рабочие кривые обычно становятся нелинейными. Это обусловлено ограниченными возможностями используемых методов анализа и оборудования. Если концентрация определяемого вещества попадает в нелинейную область высоких значений, то образец следует разбавить и анализ повторить. Чувствительность метода характеризуется величиной изменения измеряемого параметра при данном изменении концентрации. Она равна угловому коэффициенту (тангенсу угла наклона) рабочей кривой. Как правило, чем выше чувствительность, тем надежнее результаты и тем ниже порог обнаружения. Результат измерения часто включает составляющую, не связанную с определяемым веществом, - ее называют фоном. Наличие фона может быть связано с особенностями оборудования или влиянием матрицы, в которую включен образец (см. ниже). Чтобы оценить величину фона, проводят контрольный опыт. Для этого готовят контрольный образец, в котором нет определяемого вещества, а есть только все посторонние примеси, имеющиеся в матрице, а также реагенты, добавляемые в процессе анализа. Контрольный образец подвергают той же аналитической процедуре, что и определяемое вещество. Значение измеряемого параметра для этого контрольного образца считают равным фону. Порог обнаружения - это наименьшая концентрация определяемого вещества, при которой сигнал заметно отличается от фона. Величина порога обнаружения зависит от чувствительности и точности метода: чем они выше, тем ниже минимальные определяемые концентрации. Химики-аналитики систематически разрабатывают способы измерения все более низких концентраций. Сегодня для многих методов анализа порог обнаружения составляет 10-6 - 10-9 М, а некоторые недавно разработанные методы позволяют измерять пикомолярные концентрации (ниже 10-12 М), обнаруживать вещества в абсолютных количествах менее 10-18 молей (приблизительно несколько сотен тысяч молекул) и даже наблюдать отдельные атомы
(см. также ЭЛЕКТРОННЫЙ МИКРОСКОП).
Одна из задач, которые постоянно приходится решать в аналитической химии, - совершенствование методов, позволяющее работать со все более мелкими образцами. Те методы, для которых когда-то требовались миллилитровые количества, теперь обходятся микролитрами, а некоторые - и десятками пиколитров.
Матрица. Термин "матрица" относится к окружению определяемого вещества. Это все вещества, присутствующие в образце, в т.ч. и определяемые, отличные от данного. Так, хлор определяют в плазме крови, консервированной моркови, питьевой или морской воде. Эти образцы различаются по своим химическим и физическим свойствам, а следовательно, их матрицы тоже различны. Простейшая матрица - питьевая вода: она содержит относительно немного веществ, концентрация которых к тому же невелика. Консервированная морковь - сложная матрица, главным образом потому, что в ней содержатся разные органические соединения. Стандарты и определяемые при анализе вещества по возможности должны находиться в одинаковых или сравнимых матрицах, однако получить калиброванные матрицы удается очень редко. Чтобы решить эту проблему, используют синтетические матрицы, метод внутреннего стандарта и т.д. Если матрица данного образца обладает относительно постоянными физическими и химическими свойствами, не зависящими от того, когда и где был получен образец, то ее можно достаточно полно охарактеризовать и воспроизвести. Одна из таких матриц - морская вода. Концентрации ее основных компонентов (Na, Mg, Cl...) хорошо известны. Можно получить искусственную морскую воду и использовать ее для приготовления стандартных растворов других веществ, концентрация которых невелика (например, Al, Au, Ni, Zn). Состав биологических жидкостей, таких, как плазма крови или моча, также известен, что позволяет создавать искусственные матрицы для проведения определенных анализов. Другой метод состоит в том, что для стандартов и исследуемого вещества создают матрицы примерно одинакового состава. Для этого к образцу и стандартам добавляют большое количество какого-либо "инертного" вещества (для получения растворов одинаковой ионной силы к образцу и стандартам можно добавить 1 М NaClO4), так что небольшие различия в других компонентах матрицы становятся несущественными. Влияние матрицы при этом не исключается, напротив, оно усиливается, но теперь это влияние в исследуемом образце и стандарте практически одинаково. Удобный способ компенсации влияния матрицы, а также решения проблем, связанных с потерями вещества в ходе сложного анализа, - использование внутреннего стандарта. Метод состоит в следующем. Перед тем как определять вещество A, к содержащему его образцу добавляют известное количество вещества B. Количества A и B определяют по одной и той же методике. Установив соотношение между найденным и известным количествами B, корректируют полученное при анализе количество A. Внутренний стандарт должен отсутствовать в исходном образце и представлять собой химический аналог определяемого вещества. Например, для определения натрия в плазме крови методом пламенной эмиссионной спектроскопии в качестве внутреннего стандарта часто используют литий, поскольку он химически аналогичен натрию и в крови обычно отсутствует. В методе добавленных стандартов для приготовления стандартов сравнения используют сам исследуемый образец. Предположим, что мы хотим определить содержание натрия в плазме крови. Исходный образец делят на несколько частей, например на три. К одной из них ничего не добавляют, к двум другим добавляют известные количества определяемого вещества (в данном случае Na), так что его концентрация становится на 100 и 200 ммоль/л больше, чем в исходном образце. Далее по одной и той же методике определяют Na во всех частях образца и строят график зависимости измеряемой величины от прироста концентрации. Из графика определяют концентрацию натрия в исходном образце.
Равновесные и кинетические измерения. На рис. 2 графически представлен ход химической реакции Определяемое вещество + Реагенты Продукты Вначале концентрация линейно меняется во времени, затем изменение становится все более медленным, и, наконец, концентрация выходит на горизонталь - система достигает равновесия.
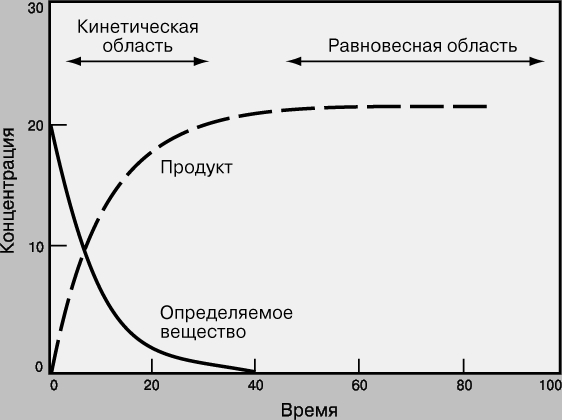
Рис. 2. ХОД ХИМИЧЕСКОЙ РЕАКЦИИ, представленный в виде графика зависимости концентрации реагента от времени.
Хотя считается, что многие химические реакции идут "до конца" (до полного исчерпания исходных веществ), на самом деле ни одна из них не протекает только в одном направлении. Химическая реакция идет до тех пор, пока концентрация всех участвующих в ней веществ не перестает меняться, т.е. пока система не достигнет равновесия. В этом состоянии концентрация некоторых веществ может быть очень мала, но все же она не равна нулю. Реакция не прекращается, просто скорость прямой реакции (Реагенты (r) Продукты) становится равной скорости обратной (Продукты (r) Реагенты), при этом происходит быстрое взаимное превращение реагентов и продуктов реакции, так что никакого суммарного изменения концентраций нет.
См. ХИМИЧЕСКАЯ КИНЕТИКА И ХИМИЧЕСКОЕ РАВНОВЕСИЕ. Аналитические определения можно проводить в химических системах, находящихся как в равновесном, так и в неравновесном состояниях. В первом случае концентрации веществ не меняются, поэтому продолжительность анализа несущественна и не влияет на выбор методики. Равновесные концентрации веществ связаны друг с другом через константу равновесия. Для химической реакции aA + bB cC + dD эта константа равна

где в квадратных скобках указаны молярные концентрации соответствующих веществ, а показатели степени равны стехиометрическим коэффициентам уравнения химической реакции. Константы равновесия реакций, используемых в аналитической химии, изменяются от 1 до 10 100 и более. Многие методы анализа основаны на определении состояния равновесия. В качестве примера можно привести рассматриваемые ниже классические методы - гравиметрию и титрование. При анализе неравновесных систем определяют изменение концентрации реагирующих веществ во времени, т.е. скорость реакции. Она задается выражением

где k - константа скорости, [[A]], [[B]], [[C]] - молярные концентрации веществ A, B, C, сумма x, y, z - порядок реакции. Какие именно химические вещества фигурируют в уравнении скорости такой реакции и с какими степенями входят их концентрации, зависит от механизма химической реакции. При неравновесных определениях нужно успеть провести измерения за достаточно малое (по сравнению с продолжительностью самой реакции) время, так чтобы концентрации реагентов не изменились. Определения, основанные на измерении скорости реакции, могут быть выполнены в очень короткое время от начала реакции (иногда несколько секунд), поскольку не нужно ждать, когда система достигнет равновесия. Если определяемое вещество - катализатор, то измерения следует проводить до достижения равновесия, поскольку катализатор изменяет только скорость реакции, но не положение равновесия. Применение кинетических измерений в аналитической химии не ограничивается методами анализа, основанными на измерении скорости реакции. В основе многих аналитических методов, например флуоресцентного анализа, амперометрии, хроматографии, лежат кинетические процессы, хотя анализируются системы, находящиеся в состоянии равновесия. Ниже описаны некоторые распространенные методы анализа.
МЕТОДЫ АНАЛИЗА, ОСНОВАННЫЕ НА ОПРЕДЕЛЕНИИ ПОЛОЖЕНИЯ РАВНОВЕСИЯ
В аналитической химии есть несколько методов, основанных на определении положения химического равновесия. К ним относятся, в частности, классические гравиметрия и титриметрия, а также сравнительно новый иммунологический анализ.
Гравиметрия (весовой метод). В гравиметрии определяемое вещество переводят в химически чистое состояние или превращают в весовую форму - соединение с точно известным постоянным составом, которое можно легко выделить и взвесить. Количество анализируемого вещества рассчитывают исходя из массы весовой формы и уравнения реакции, связывающей это вещество с весовой формой. Химические стандарты не требуются. Весовые методы анализа очень точны, их часто используют в сомнительных случаях в качестве контроля. Точность анализа ограничивается точностью определения массы и полнотой образования и выделения чистого вещества. Гравиметрия - продолжительная процедура, поскольку и перевод определяемого вещества в весовую форму, и выделение ее из смеси требуют времени. Кроме того, необходимо убедиться в том, что весовая форма - это вещество точно известного постоянного состава, не содержащее примесей. Большинство весовых определений основано на образовании и выделении из раствора (как правило, водного) твердых нерастворимых осадков. Задача состоит в том, чтобы осадить по возможности максимальное количество определяемого вещества (по крайней мере, 99,99%), поэтому осадок (чаще всего соль) должен обладать как можно меньшей растворимостью. Растворимость соли определяется величиной константы равновесия реакции растворения, в которой образуются ионы. Количественное осаждение обычно осуществляют, добавляя к раствору с определяемым веществом стехиометрический избыток осаждающего реагента. Растворимость соли в присутствии избытка одного из ионов, входящих в ее состав, снижается. Для уменьшения влияния других равновесных реакций, приводящих к увеличению растворимости соли, необходимо контролировать состав раствора. Для разных анализируемых веществ применяют разные осаждающие реагенты. Некоторые из них приведены в табл. 1.
Одно из основных преимуществ весовых определений заключается в том, что не нужно калибровать приборы или готовить стандартные растворы. Результат получают, взвесив осадок и зная состав участвующих в реакции соединений. Пусть, например, мы хотим определить содержание марганца в образце. Для этого нужно перевести марганец в Mn3O4, отделить последний и взвесить. Предположим, что из 1,52 г образца образуется 0,126 г Mn3O4 (т.е. 0,00055 моль, так как 1 моль Mn3O4 содержит 228,8 г). В 1 моль Mn3O4 содержится 3 моль Mn, а в 0,00055 моль - соответственно 0,00165 моль Mn, или 0,0907 г (1 моль Mn содержит 54,94 г). Следовательно, содержание Mn в образце (0,0907/1,52)*100% = 5,97%. Как мы уже говорили, гравиметрия довольно медленная процедура; образование осадка, его отделение фильтрованием, высушивание - все это требует времени. Кроме того, весовые определения обычно не отличаются высокой селективностью, поэтому дополнительное время уходит на подбор условий (например, pH), переосаждение и т.п. Чем сложнее по составу образец, тем более вероятны ошибки: завышение весового содержания анализируемого вещества, связанное с соосаждением примесей, или занижение, обусловленное потерей вещества на стадии его выделения. Вследствие ограниченных селективности и чувствительности гравиметрию нет смысла применять, когда в наличии имеются лишь микро- или следовые количества определяемого вещества.
Титриметрия (объемный метод). В титриметрии концентрацию определяют, измеряя объем стандартного или титрованного реагента (титранта), израсходованного в химической реакции с определяемым веществом в растворе (или газовой фазе). Измерение проводят с помощью процедуры титрования. Это простой, относительно быстрый, универсальный и точный метод. При титровании титрант добавляют порциями или непрерывно с небольшой постоянной скоростью и измеряют его объем до тех пор, пока не будет достигнута точка эквивалентности, отвечающая объему титранта, при котором в реакцию вступает все определяемое вещество. Точку эквивалентности находят, непрерывно следя за изменением тех или иных свойств титруемого раствора (цвета, оптической плотности, электрохимических свойств и т.д.) при помощи специальных приборов или визуально. Чтобы данную химическую реакцию можно было использовать в титровании, участвующие в ней вещества должны находиться в строго определенных количественных (стехиометрических) соотношениях. Реакция должна протекать быстро и практически до конца, а точка эквивалентности точно фиксироваться. Чаще всего используют реакции нейтрализации (кислотно-основные), комплексообразования и окислительно-восстановительные. Реакции нейтрализации распространены наиболее широко; именно их мы и рассмотрим для пояснения ключевых моментов всех реакций титрования.
Кривые титрования. Кривая титрования - это график зависимости pH, оптической плотности или каких-либо других характеристик титруемого раствора (ось ординат) от объема добавленного титранта (ось абсцисс). Масштаб оси абсцисс всегда линейный, а оси ординат может быть линейным или логарифмическим. Линейный масштаб удобен для тех методов контроля за титрованием (спектрофотометрия, амперометрия), в которых контролируемый параметр меняется с концентрацией линейно, а логарифмический - в случае логарифмического изменения (например, при потенциометрии с ионоселективным электродом). Логарифмический масштаб часто используют при визуальном определении конечной точки титрования, поскольку именно в этом масштабе наиболее наглядно проявляется резкое изменение свойств раствора вблизи точки эквивалентности. Зависимость кривых титрования от концентрации и константы равновесия. Для точного определения конечной точки титрования необходимо, чтобы на кривой титрования вблизи точки эквивалентности наблюдался перегиб (скачок). Это требование устанавливает пределы как для минимальной определяемой концентрации, так и для минимальной константы равновесия, приемлемой для реакции титрования. На рис. 3 представлены кривые титрования сильной кислоты сильным основанием и слабой кислоты сильным основанием. Видно, что при уменьшении концентрации скачок становится менее выраженным. Нижний предел концентрации зависит от конкретной реакции и метода определения конечной точки титрования, но проводить титрование при концентрациях ниже 10-4 М уже затруднительно. Рисунок 4 иллюстрирует влияние константы равновесия реакции титрования на кривую титрования. Для реакций нейтрализации в водных растворах константа равновесия в случае сильной кислоты и сильного основания составляет 1014, а для слабой кислоты и сильного основания - 10 14Ka, где Ka - константа диссоциации кислоты. По мере уменьшения константы равновесия уменьшается и величина скачка. Чтобы визуальное определение конечной точки титрования было надежным, константа равновесия не должна быть меньше 106. При инструментальном контроле титрования или расчете положения конечной точки титрования на основании полученных данных константа равновесия может составлять всего 10 2.
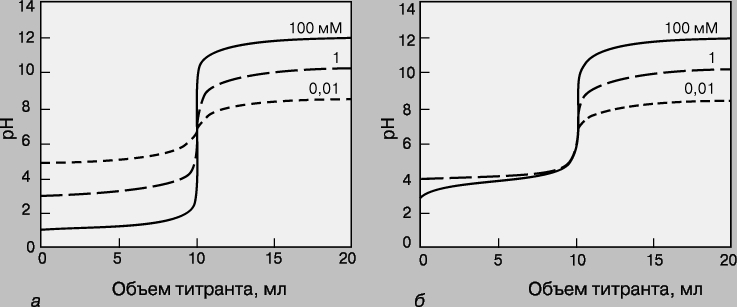
Рис. 3. КРИВЫЕ ТИТРОВАНИЯ сильной кислоты сильным основанием (а) и слабой кислоты (pKa = 5) сильным основанием (б) для разных концентраций определяемого вещества.
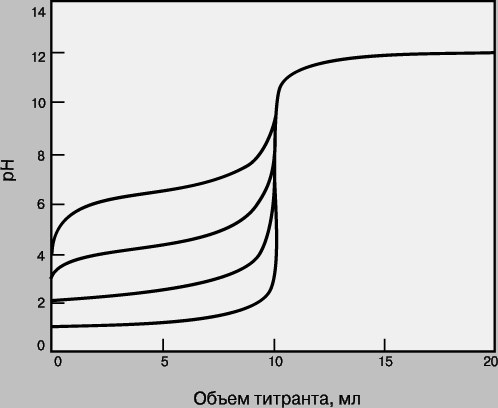
Рис. 4. КРИВЫЕ ТИТРОВАНИЯ ПРИ РАЗНЫХ КОНСТАНТАХ РАВНОВЕСИЯ. Титруемые кислоты (снизу вверх): сильная кислота типа HCl (Kр = 1014); кислота с pKa = 3 (Kр = 1011); кислота с pKa = 5 (Kр = 109); кислота с pKa = 7 (Kр = 107).
Смеси. Если в образце содержатся два определяемых вещества, взаимодействующие с одним и тем же титрантом, их можно определить в одной операции титрования при условии, что реакция каждого из этих веществ с титрантом имеет достаточно высокую константу равновесия и что эти константы существенно различаются (как правило, не менее чем на два порядка). На рис. 5 приведены кривые титрования для смесей анализируемых веществ с различными pKa. Первым титруется вещество, реакция которого с титрантом имеет большую константу равновесия. Объем от начала титрования до первой конечной точки позволяет определить концентрацию этого анализируемого вещества, а объем между конечными точками титрования - концентрацию второго анализируемого вещества. Если константы равновесия слишком близки, то локализовать первую конечную точку титрования будет сложно или вообще невозможно. В таком случае анализируемые вещества нельзя определить по отдельности, а суммарный объем титранта позволит рассчитать лишь сумму их концентраций.
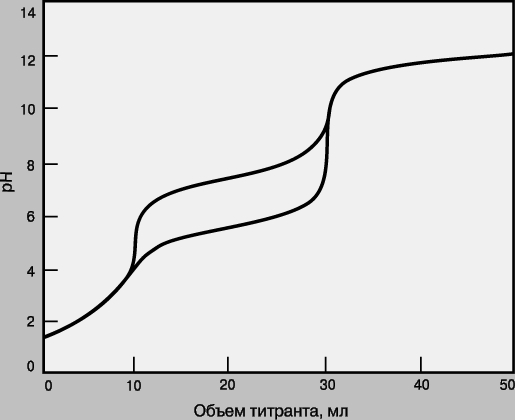
Рис. 5. КРИВЫЕ ТИТРОВАНИЯ для смеси двух определяемых веществ с pKa = 2 (Kр = 10 12) и pKa = 6 или 8 (Kр = 10 8 или 10 6).
Цветные индикаторы. Цветной индикатор - вещество, которое меняет свою окраску при взаимодействии с одним из компонентов титруемого раствора. Пусть, например, индикатор In взаимодействует с определяемым веществом A:
A + T P (реакция титрования) A + In AIn (реакция с индикатором)
где A - определяемое вещество, T - титрант, P - продукт реакции, In - индикатор, AIn - продукт взаимодействия определяемого вещества и индикатора. In можно использовать как индикатор в данной реакции титрования, если он и AIn по-разному окрашены. В качестве цветных индикаторов обычно используют вещества, вступающие в реакции того же типа, что и реакция между определяемым веществом и титрантом. Индикаторами кислотно-основного титрования, как правило, являются слабые кислоты, у которых кислая и основная формы имеют разную окраску. В реакциях комплексообразования индикаторами служат вещества, способные образовывать комплексы с определяемым ионом металла и имеющие разную окраску в зависимости от того, находятся ли они в свободном состоянии или входят в состав комплекса. Очень часто при титровании меняется pH раствора (кислотно-основное титрование) или потенциал (окислительно-восстановительные реакции). Для кислотно-основного титрования используют индикатор, который переходит из одной окрашенной формы в другую при pH, близком к pH точки эквивалентности. На рис. 6 представлена кривая титрования смеси двух слабых кислот и указаны интервалы изменения окраски нескольких индикаторов. Для фиксации первой конечной точки титрования следует взять бромкрезоловый зеленый, второй - тимолфталеин.
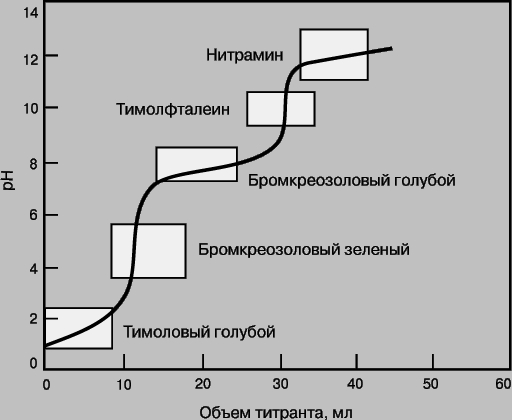
Рис. 6. КРИВАЯ ТИТРОВАНИЯ СМЕСИ ДВУХ СЛАБЫХ КИСЛОТ с pKa = 2 и 7. Указаны интервалы изменения окраски индикаторов в соответствующих диапазонах pH.
Инструментальное определение конечной точки титрования. Непрерывный контроль процесса титрования с помощью приборов позволяет получить данные о его ходе как до, так и после точки эквивалентности. Эти данные можно представить в виде графика и определять конечную точку графически или путем вычислений. Чаще всего используют спектрофотометрическое (измерение оптической плотности), амперометрическое и потенциометрическое (измерение электродного потенциала) титрование.
Кулонометрическое титрование. Кулонометрическое титрование обычно проводят при постоянном токе. Титрант образуется в результате электрохимических процессов на рабочем электроде в сосуде для титрования. Число молей анализируемого вещества равно произведению силы тока на время, необходимое для образования титранта в количестве, достаточном для достижения конечной точки титрования с учетом стехиометрии. Химические стандарты не требуются. К титрантам, которые образуются в ходе электрохимических процессов, относятся H+, OH-, Br2 и I2.
Прямое и обратное титрование. В простейшем варианте титрования анализируемое вещество взаимодействует непосредственно с титрантом. Количество анализируемого вещества рассчитывают исходя из молярной концентрации титранта, его объема, требуемого для достижения точки эквивалентности, и стехиометрии реакции между определяемым веществом и титрантом. Предположим, что для достижения конечной точки титрования 5,00 мл раствора, содержащего ионы Sn2+, потребовалось 12,51 мл 0,100 М раствора Ce(IV). Реакция титрования имеет вид Sn2+ + 2Ce4+ -> Sn4+ + 2Ce3+. Количество Ce4+, пошедшего на титрование, составляет (12,51*10-3 л)*(0,100 моль/л) = 12,51*10-4 моль, количество прореагировавшего Sn2+ в 2 раза меньше, т.е. 6,25*10-4 моль. Столько Sn2+ содержится в 5,00 мл раствора, так что его концентрация равна
(6,25*10-4 моль)/(5*10-3 л) = 0,125 М. В обратном титровании анализируемое вещество взаимодействует не с титрантом, а с другим реагентом, присутствующим в избытке. Избыток затем определяют титрованием. Если известно исходное количество реагента и определен его избыток, то разность между ними - это количество реагента, пошедшее на реакцию с определяемым веществом. Предположим, что к 5,00 мл образца, содержащего фенол, добавляют 20,00 мл 0,100 М раствора гидроксида натрия. В результате реакции образуется фенолят натрия. Избыток гидроксида натрия титруют 12,53 мл 0,0800 М раствора HCl. Соотношения между реагентами в реакциях гидроксида натрия и фенола или гидроксида натрия и соляной кислоты составляют 1:1. В таком случае исходное количество гидроксида натрия равно (20,00Ч10-3 л)*(0,100 моль/л) = 20,00*10-4 моль. Избыток гидроксида натрия равен количеству соляной кислоты, пошедшей на его титрование: (12,53Ч10-3 л)*(0,0800 моль/л) = 10,00*10-4 моль. На взаимодействие с анализируемым веществом израсходовано (20,00 - 10,00)*10-4 моль = 10,00*10-4 моль гидроксида натрия. Такое же количество фенола содержится в 5,00 мл образца. Следовательно, концентрация фенола составляет (10,00*10-4 моль)/(5,00*10-3 л) = 0,200 М. Обратное титрование используют, например, когда константа равновесия реакции прямого титрования слишком мала. Так, в рассмотренном выше примере фенол - довольно слабая кислота, и константа равновесия прямого титрования фенола гидроксидом натрия составляет величину лишь порядка 104. В то же время константа равновесия реакции обратного титрования между избытком гидроксида натрия (сильное основание) и соляной кислотой (сильная кислота) равна 1014. Среди других причин применения обратного титрования - отсутствие подходящего метода индикации или недостаточная скорость реакции при прямом титровании. Так, для прямого комплексонометрического титрования иона металла этилендиаминтетрауксусной кислотой (ЭДТА) обычно используют металлы-индикаторы. Понятно, что для определения конечных точек титрования всех ионов металлов нужно множество различных индикаторов. При обратном титровании к раствору, содержащему ион металла, добавляют избыточное количество ЭДТА, а избыток последнего затем определяют при помощи раствора, содержащего Mg2+. Тогда единственный необходимый индикатор - это индикатор на Mg2+, независимо от того, какой ион определяют.
Кислотно-основное титрование. Случаев применения титрования кислот и оснований множество. Чтобы конечная точка титрования определялась наиболее четко, в качестве титрантов применяют сильные кислоты и основания. Типичный кислотный титрант - HCl. Его стандартизуют по первичному стандартному карбонату натрия, используя в качестве индикаторов метиловый красный, метиловый оранжевый или бромкрезоловый зеленый. Типичный основный титрант - NaOH, его стандартизуют по первичному стандартному бифталату калия, используя в качестве индикатора фенолфталеин. Примером кислотно-основного титрования может служить метод определения содержания азота в различных соединениях (метод Кьельдаля): образец разлагают горячей серной кислотой, превращая азот в ион аммония; после охлаждения образец обрабатывают щелочью, чтобы перевести ион аммония в аммиак; аммиак улавливают кислым раствором, после чего избыток кислоты определяют титриметрически при помощи реакции нейтрализации.
Комплексонометрическое титрование. Чаще всего комплексонометрическое титрование применяют для определения ионов металлов с использованием ЭДТА в качестве титранта (например, при определении жесткости воды). Образец воды подщелачивают аммиачным буферным раствором, добавляют индикатор эриохром черный и полученный раствор титруют ЭДТА.
Окислительно-восстановительное титрование. Во многих наиболее распространенных реакциях окислительно-восстановительного титрования косвенным участником является иод. Конечная стадия титрования заключается в количественном определении иода при помощи титрования тиосульфатом натрия. В качестве индикатора на иод используют крахмал. Тиосульфат стандартизуют по трииодид-иону (I3-), который получается по реакции между KI и первичным стандартом KIO3. Таким способом определяют, например, степень ненасыщенности жирных кислот, содержание фенола, многоатомных спиртов (глицерина или этиленгликоля).
СПЕКТРОСКОПИЯ
Спектроскопические методы основаны на взаимодействии электромагнитного излучения с веществом, т.е. на определении характеристик поглощаемого, испускаемого или рассеянного излучения. Часто параллельно со спектроскопическими методами используют масс-спектрометрию; хотя с ее помощью и не изучают взаимодействие излучения с веществом, результаты измерений обычно представляют в виде спектра.
Основные положения. Электромагнитное излучение характеризуется энергией E, частотой n и длиной волны l, которые связаны между собой соотношением E = hn = hc/l, где h - постоянная Планка (6,63Ч10-34 Дж*с), c - скорость света (3*108 м/с). Под действием электромагнитного излучения молекулы вещества переходят на более высокие энергетические уровни. На рис. 7 схематически представлены эти уровни (горизонтальные линии) и некоторые переходы между ними (вертикальные стрелки). Энергия поглощается и испускается дискретными порциями (квантами), и чтобы поглощение произошло, энергия кванта падающего излучения должна в точности соответствовать энергии перехода в одно из возбужденных состояний поглощающей молекулы. Когда молекула из возбужденного состояния переходит на более низкий энергетический уровень, излучение испускается, при этом энергия излучения равна разности энергий двух уровней. Спектр - это зависимость интенсивности поглощения или испускания электромагнитного излучения от длины волны или энергии. Он состоит из пиков или полос разной высоты. Положение пика относительно оси абсцисс (l или E) указывает разность в энергии двух уровней. Характер спектра дает информацию о природе поглощающего или испускающего вещества, а высота пиков - о числе молекул, участвующих в переходе (т.е. о концентрации вещества).
См. также СПЕКТР; СПЕКТРОСКОПИЯ.

Рис. 7. ЭНЕРГЕТИЧЕСКИЕ УРОВНИ гипотетической молекулы. E - электронные уровни, v - колебательные. Сплошные стрелки отвечают поглощению или испусканию света, пунктирные - безызлучательным переходам.
Диапазон энергий всего электромагнитного спектра очень широк. В табл. 2 приведены общепринятые названия типов излучения, указаны диапазоны их энергий и длин волн, а также типы переходов.
Методам, чаще всего используемым в аналитической химии, соответствуют ультрафиолетовая (УФ), видимая, инфракрасная (ИК) области и радиоволны. На рис. 8 представлены блок-схемы устройств для получения спектров поглощения и испускания.
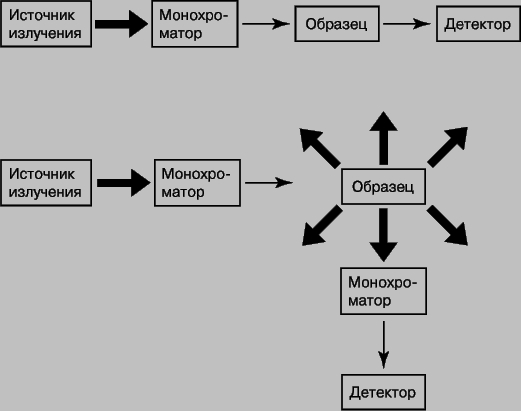
Рис. 8. БЛОК-СХЕМЫ устройств для получения спектров поглощения (вверху) и испускания (внизу). Жирные стрелки соответствуют полихроматическому излучению, тонкие - монохроматическому.
Выбор конкретного оборудования - источника излучения, монохроматора, детектора - зависит от длин волн используемого излучения и характера измерений. В УФ- и видимой областях в качестве источников света обычно используют лампы накаливания или лазеры, монохроматором служит щель или призма, а детектором - фотоэлектронный умножитель и блок фотодиодов. Поглощение в УФ- и видимой областях. Спектры поглощения в УФ- и видимой областях содержат как качественную, так и количественную информацию о поглощающем веществе. Последнее и позволяет использовать их в аналитической химии. Поглощение света подчиняется закону Ламберта - Бера

где D - оптическая плотность, I0 и I - интенсивности падающего и прошедшего через образец света, T - пропускание, e - молярный коэффициент экстинкции, l - длина оптического пути (толщина поглощающего слоя) в см, c - молярная концентрация. Измерив оптическую плотность D, из соотношения D = ecl можно найти концентрацию поглощающего вещества. Образцы, используемые в абсорбционной спектроскопии в УФ- и видимой областях, - это, как правило, разбавленные растворы. Диапазон концентраций, которые можно определить, зависит от молярного коэффициента экстинкции исследуемого вещества, максимальное значение которого составляет ХИМИЯ АНАЛИТИЧЕСКАЯ105 (отметим, что измерения следует проводить при длине волны, соответствующей максимуму в спектре поглощения). Для получения достоверных результатов измеряемая оптическая плотность должна находиться в диапазоне 0,01-2. При толщине поглощающего слоя в 1 см это соответствует концентрации 10-8 М, что в 1000 раз ниже, чем при титровании. Обычно в рабочей области (области линейности) измерений концентрация может изменяться по меньшей мере в 100 раз. Селективно подбирая длину волны, отвечающую максимуму поглощения вещества, можно исключить влияние матрицы (растворителя). Измерения оптической плотности непродолжительны, что позволяет определять с их помощью скорости реакций. Если исследуется смесь нескольких поглощающих веществ, то концентрацию каждого из них определяют, проводя измерения при длинах волн, отвечающих максимумам поглощения этих веществ.
Люминесценция. В люминесцентной спектроскопии измеряется интенсивность излучения, испускаемого атомами или молекулами вещества при их переходе из возбужденного состояния в основное (рис. 8). Люминесценция бывает двух типов: флуоресценция и фосфоресценция. При флуоресценции атом или молекула переходит в основное состояние из короткоживущего возбужденного состояния. Она наблюдается почти сразу после поглощения, быстро спадает и исчезает в результате столкновений излучающей молекулы с другими молекулами в растворе (тушение флуоресценции). Фосфоресценция наблюдается при переходе молекулы в основное состояние из относительно долгоживущего возбужденного состояния, так что между поглощением света и испусканием может пройти относительно много времени. Для фосфоресценции характерны большая длина волны излучения, меньшая высота пиков и большее влияние матрицы. Флуоресцентные измерения более избирательны, чем спектрофотометрические, поскольку зависят сразу от двух длин волн: поглощаемого и испускаемого света. Интенсивность флуоресценции связана с интенсивностью поглощенного света следующим соотношением: Iисп = kIпогл. Это соотношение линейно относительно концентрации только при малых ее значениях: Iисп = kўIпоглC. Здесь k и kў - константы, характеризующие свойства молекулы, связанные с поглощением и испусканием излучения, а C - концентрация определяемого вещества. Флуоресцентный анализ позволяет измерять в 1000 раз меньшие концентрации, чем спектрофотометрический. Это связано с характером сигнала, определяемого в том и другом случаях: во флуоресцентных измерениях нужно зарегистрировать небольшую разницу между двумя слабыми сигналами, а при измерениях поглощения - между сильными, что гораздо сложнее. Если свет испускается в результате химической реакции, то процесс называют хемилюминесценцией. Интенсивность излучения зависит от скорости химической реакции, а последняя, в свою очередь, от концентрации. Таким образом, измеряя интенсивность хемилюминесценции, можно определить концентрацию соответствующего реагента. В качестве примера люминесцентного определения приведем реакцию с участием люминола. При окислении пероксидом водорода в присутствии комплексов переходных металлов это вещество люминесцирует, что позволяет проводить количественное определение следовых количеств ионов металлов (или некоторых комплексов), а также пероксида водорода.
Инфракрасная (ИК) спектроскопия. Спектры поглощения в видимой и УФ-областях, о которых шла речь выше, возникают в результате электронных переходов в атомах и молекулах. Поглощение же в ИК-области обусловлено переходами между колебательными уровнями, отвечающими разной колебательной энергии функциональных групп. В ИК-спектроскопии чаще всего используют среднюю часть ИК-области, 4000-200 см-1. Для интерпретации ИК-спектров составлены специальные каталоги и таблицы, в которых указаны характеристические частоты колебаний различных групп (табл. 3).
Таблица 3. ХАРАКТЕРИСТИЧЕСКИЕ ЧАСТОТЫ КОЛЕБАНИЙ НЕКОТОРЫХ ГРУПП
Группа (тип колебаний) Волновое число, см-1
O-H (валентные) 3350-3250 N-H (валентные) 3460-3280 C-H (валентные) 2980-2850 Cє C (валентные) 2300-2100 C=O (валентные) 1870-1650 C=N (валентные) 1620-1560 C=C (валентные) 1645-1615 N-H (деформационные) 1650-1590 C-H (деформационные) 1470-1360 O-H (деформационные) 1440-1260
Значения молярных коэффициентов экстинкции для ИК-области меньше, чем для видимой и УФ-областей, поэтому с помощью ИК-спектроскопии можно исследовать или чистые вещества, или очень концентрированные растворы. Жидкости заливают между оптически прозрачными стеклами, где они образуют тонкую пленку, или в кювету, твердые вещества измельчают и суспендируют в оптически прозрачной среде. Растворы исследовать сложнее, чем твердые вещества, поскольку растворитель часто поглощает в этой же области. Чтобы повысить чувствительность и разрешающую способность метода, в современных модификациях используют ИК-спектроскопию с фурье-преобразованием.
Ядерный магнитный резонанс (ЯМР). Метод ЯМР основан на резонансном поглощении электромагнитной энергии, обусловленном магнетизмом ядер. Это поглощение наблюдается в сильном магнитном поле, под действием которого энергетические уровни ядер, обладающих магнитным моментом, расщепляются. Наложение небольшого по величине и изменяющегося по частоте электромагнитного поля вызывает переходы между уровнями, проявляющиеся в виде линий поглощения в спектрах ЯМР. Метод ЯМР - один из наиболее эффективных методов структурных исследований. Он позволяет получить информацию о строении молекул, о том, какие ядра присутствуют в определяемом веществе и в каком количестве, каково их окружение. Один из вариантов ЯМР - протонный магнитный резонанс (1H-ЯМР) - часто оказывается единственным методом, позволяющим определить строение органических соединений. Вслед за ним иногда применяют 13C-ЯМР, масс-спектрометрию и ИК-спектроскопию. Чаще всего методом ЯМР определяют ядра атомов водорода (протоны, 1H) и 13C. Можно также изучать другие ядра, например 19F, 31P, 17O, 15N и 29Si. Спектр ЯМР представляет собой зависимость интенсивности поглощения от относительной величины d (химический сдвиг), определяемой как

где H и n - напряженность магнитного поля и резонансная частота для образца и стандарта. Значения d даются в миллионных долях (м.д.; в англоязычной литературе - ppm, parts per million). В качестве стандарта в случае 1H и 13C обычно используют тетраметилсилан (ТМС). Наиболее важными характеристиками ЯМР-спектра являются положение сигналов (полос) поглощения, их интенсивность и мультиплетность. Поскольку электроны частично экранируют ядро и изменяют величину действующего на него магнитного поля, положение сигналов поглощения различных ядер (например, протонов) зависит от их электронного окружения. В группах H-N-, H-O- и H-C - протоны поглощают при разных частотах и имеют разный химический сдвиг. Значения измеренных химических сдвигов для большого числа структурных элементов сведены в таблицы. Спин-спиновое взаимодействие протонов соседних атомов приводит к расщеплению сигнала ЯМР, при этом мультиплетность сигнала зависит от числа участвующих во взаимодействии протонов. Перекрывание и расщепление сигналов существенно усложняют ЯМР-спектры. Для их упрощения используют более сильные магнитные поля, что позволяет растянуть спектр и уменьшить перекрывание пиков. На рис. 9 приведен ЯМР-спектр диэтилового эфира CH3CH2OCH2CH3, иллюстрирующий представления о химическом сдвиге и расщеплении. Спектр показывает, что в молекуле присутствуют протоны двух видов. Триплет с d = 1,1 м.д. - это сигнал от протонов метильной группы CH3-, а квартет с d = 3,3 м.д. - сигнал от протонов метиленовой группы -CH2-.
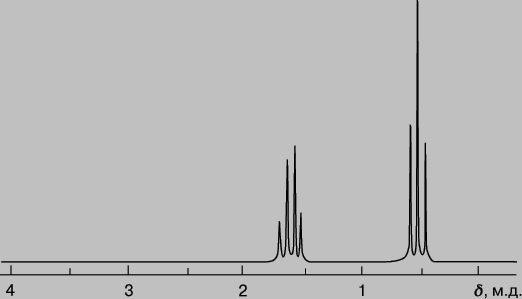
Рис. 9. СПЕКТР ПРОТОННОГО МАГНИТНОГО РЕЗОНАНСА диэтилового эфира.
Для получения спектров более высокого разрешения и для ускорения процедуры в ЯМР-спектроскопии используют преобразование Фурье.
Масс-спектрометрия (МС). Масс-спектрометрия - один из наиболее эффективных и широко применяющихся аналитических методов. Его отличают высокая селективность, чувствительность и точность. Принцип метода состоит в том, что определяемое вещество переводят в газообразное состояние, ионизируют и образовавшиеся ионы (заряженные фрагменты исходных молекул) разделяют в магнитном поле по величинам отношения массы к заряду. Любой масс-спектрометр состоит из системы напуска образца, ионизационной камеры и системы разделения ионов. В приборе поддерживают высокий вакуум (ХИМИЯ АНАЛИТИЧЕСКАЯ10-6 мм рт.ст.). Методы регистрации отработаны настолько хорошо, что позволяют без труда производить подсчет отдельных ионов. Масс-спектр представляет собой зависимость интенсивности сигнала от отношения массы образующихся при ионизации частиц к их заряду (m/e). Спектр состоит из одного или нескольких пиков. При низкой энергии ионизирующих электронов от молекул вещества отрывается по одному электрону и образуются молекулярные ионы (M+); в этом случае в спектре присутствует единственный пик, и определить мол. массу анализируемого вещества несложно. Если энергия ионизации повышается, то молекулярный ион распадается на более мелкие осколочные ионы, которые могут участвовать затем в реакциях перегруппировки с образованием других ионов. Рассматривая энергетику и механизм этих реакций, можно по масс-спектрам воссоздать структуру исходного соединения. Следовательно, масс-спектр является своего рода "отпечатком" вещества. На рис. 10 представлены масс-спектры 2-метилбутана и 2,2-диметилпропана (неопентана) - насыщенных углеводородов с эмпирической формулой C5H12. Ионизацию, которая приводит к значительной фрагментации, называют жесткой. В отличие от нее при мягкой ионизации наблюдается значительно меньшая фрагментация, но увеличивается высота пика молекулярного иона. В качестве примера на рис. 11 представлены масс-спектры мефобарбитала (одного из барбитуратов), полученные в результате ионизации электронным ударом, химической ионизации и полевой десорбции.
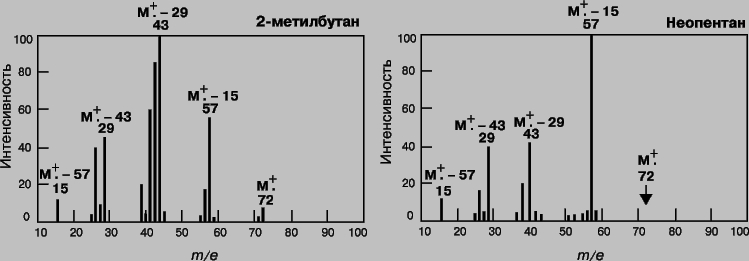
Рис. 10. МАСС-СПЕКТРЫ 2-метилбутана и неопентана (ионизация электронным ударом).
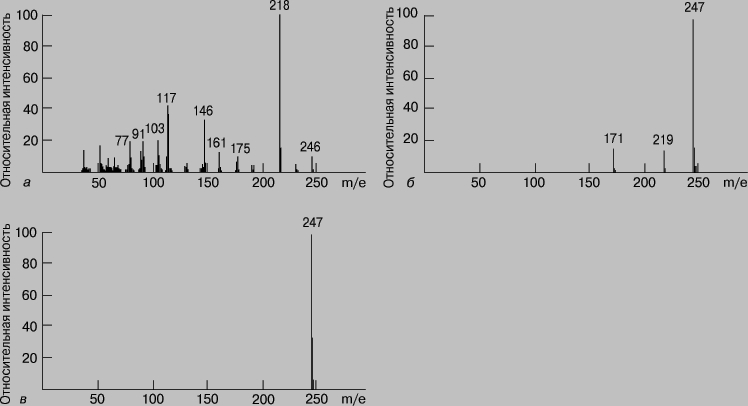
Рис. 11. МАСС-СПЕКТРЫ МЕФОБАРБИТАЛА при ионизации электронным ударом (а), химической ионизации (б) и десорбции полем (в).
Способы ионизации. Выбор способа ионизации зависит от свойств определяемого вещества, матрицы, в которую оно включено, и желаемой степени ионизации. При помощи стандартного оборудования можно ионизировать вещества с мол. массой до 20 000, но есть приборы и для ионизации соединений, мол. масса которых достигает 150 000-200 000. При ионизации электронным ударом молекулы газообразного вещества бомбардируют потоком электронов, в результате чего образуется множество осколочных ионов. Масс-спектры в этом случае весьма сложны, и для их интерпретации используют специальные каталоги спектров. Один из наиболее распространенных методов мягкой ионизации летучих веществ - химическая ионизация. В этом методе в ионизационной камере поддерживают высокую концентрацию метана, который ионизируется при бомбардировке электронами в первую очередь. Затем ионы метана сталкиваются с молекулами исследуемого вещества, и в результате ион-молекулярных реакций образуются ионы (M + 1)+ и (M - 1)+. Для мягкой ионизации жидких образцов используют бомбардировку быстрыми атомами. Поток быстрых атомов ксенона или аргона бомбардирует раствор образца в глицерине, в результате чего образуется большое количество молекулярных ионов. Еще один метод мягкой ионизации - так называемая полевая десорбция; в этом случае ионы образуются под действием сильного электрического поля. Его чаще используют для ионизации неполярных, термически нестабильных веществ, а также соединений с большой мол. массой (полимеров), т.е. в тех случаях, когда нельзя применять бомбардировку быстрыми атомами. Все более широкое распространение получает ионизация распылением (электрораспыление, термораспыление и т.д.). В этом методе ввод растворенного вещества и его ионизация осуществляются в одну стадию. Для элементного и изотопного анализа применяют индуктивно-связанную аргоновую плазму. С ее помощью осуществляют отбор и ионизацию образца, а в масс-спектрометре проводят разделение ионов. Для элементного анализа поверхностей используют метод масс-спектрометрии вторичных ионов. Потоком ионов Ar+ или Xe+ бомбардируют поверхность образца, и высвободившиеся вторичные ионы направляют в масс-анализатор.
Тандемная масс-спектрометрия. В этом методе используются два последовательно соединенных масс-спектрометра. Одно из возможных применений метода - анализ смесей. Смесь определяемых веществ подвергают мягкой ионизации, в результате которой из каждого компонента образуется молекулярный ион. Ионы разделяют в первом масс-спектрометре, выполняющем функцию хроматографа. Один из компонентов вводят во второй спектрометр, где проводят его жесткую ионизацию и последующее разделение осколков. Используя каталоги, определяют структуру исходного вещества. Если предметом анализа является не смесь, а единственное вещество, последнее подвергают жесткой ионизации в первом масс-спектрометре, разделяют осколочные ионы и направляют один из них в ионизационную камеру второго прибора. Здесь осколок вновь ионизируют и подвергают последующей фрагментации. Полученный масс-спектр интерпретируют с помощью каталога и устанавливают структуру исходного осколка.
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
В основе электрохимических методов анализа лежит исследование процессов, протекающих в электролитах или на поверхности погруженных в них электродов. Эти процессы могут быть равновесными или неравновесными в зависимости от условий эксперимента и давать информацию о скорости химических реакций, природе участвующих в них соединений, термодинамике (см. также ЭЛЕКТРОХИМИЯ). Наиболее широко в аналитической химии используются следующие электрохимические методы.
Потенциометрия. В потенциометрических методах измеряется разность потенциалов между индикаторным электродом и электродом сравнения в отсутствие тока в электрохимической цепи. В этих условиях анализируемая система находится в равновесии, и электродный потенциал связан с концентрацией раствора уравнением Нернста:

где E° - стандартный потенциал окислительно-восстановительной пары ox + ne red, R - универсальная газовая постоянная, T - абсолютная температура, F - постоянная Фарадея, a - активность. При потенциометрических измерениях широко применяются ионоселективные электроды, чувствительные к какому-то одному иону (водорода, натрия, аммония). Простейший индикаторный электрод - это какой-либо благородный металл, например платина. При потенциометрическом титровании в анализируемый раствор порциями добавляют стандартный раствор реагента (см. выше Титриметрия) и следят за изменением потенциала. Получаемые S-образные кривые позволяют найти точку эквивалентности, константу равновесия, стандартный потенциал.
Вольтамперометрия. Во всех вариантах вольтамперометрических методов используют индикаторный микроэлектрод, с помощью которого получают вольтамперограммы - кривые зависимости силы тока в электрохимической ячейке от разности потенциалов. Второй, вспомогательный электрод - неполяризующийся - имеет большую поверхность, так что его потенциал практически не меняется при прохождении тока. Индикаторные электроды изготовляют в виде капилляра, из которого по каплям вытекает жидкий металл (ртуть, амальгама, галлий). Вольтамперограммы позволяют идентифицировать растворенные вещества в электролите, определять их концентрацию, а в некоторых случаях находить термодинамические и кинетические параметры. Первый вольтамперометрический метод - полярография - был предложен Я.Гейровским в 1922. Рабочим электродом в нем служил капающий ртутный электрод. Эту методику обычно применяют для определения ионов металлов (Pb2+, Cd2+, Cu2+). Среди других вольтамперометрических методов - вольтамперометрия с линейной разверткой (с монотонным изменением) потенциала, циклическая (с быстрой треугольной разверткой потенциала) вольтамперометрия. С их помощью изучают механизм электродных реакций, определяют малые концентрации веществ в растворе.
Амперометрия. В амперометрии потенциал рабочего (индикаторного) электрода поддерживают постоянным и измеряют предельный диффузионный ток в растворе. При амперометрическом титровании точку эквивалентности находят по излому кривой сила тока - объем добавленного рабочего раствора. Хроноамперометрические методы основаны на измерении зависимости силы тока от времени и применяются для определения коэффициентов диффузии и констант скорости. Электрохимические ячейки, работающие по принципу амперометрии, используются в качестве датчиков в жидкостной хроматографии.
Кондуктометрия. Этот метод основан на измерении электропроводности раствора. Условия опыта подбирают таким образом, чтобы преобладающий вклад в измеряемый потенциал ячейки вносило омическое падение напряжения IR (R - сопротивление раствора), а не скачок потенциала на границе раздела электрод/раствор. Электропроводность однокомпонентного раствора можно связать с его концентрацией, а для сложных систем оценивается общее содержание ионов в растворе. Широко используется и кондуктометрическое титрование, когда к анализируемому раствору порциями добавляют известный реагент и следят за изменением электропроводности.
Кулонометрия. В кулонометрии проводят полный электролиз раствора при контролируемом потенциале и измеряют количество электричества, необходимое для этого. Количество вещества определяют с помощью закона Фарадея P = QM/Fn, где P - масса (г) электрохимически превращенного вещества, Q - количество электричества (Кл), M - молекулярная масса вещества, F - постоянная Фарадея, n - число электронов, вовлеченных в электрохимическое превращение одной молекулы. Кулонометрические методы абсолютны, т.е. не нуждаются в калибровочных кривых. При кулоногравиметрии количество вещества, подвергшегося электролизу, определяют взвешиванием электрода до и после эксперимента.
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Обычно анализируемый образец состоит не из одного вещества, а из смеси веществ. Одни из них представляют интерес для исследователя, другие являются примесями, осложняющими анализ. И хотя существуют аналитические методики, позволяющие проводить анализ сложных смесей, всегда легче работать с чистым веществом. Для получения чистых веществ используют разнообразные методы разделения: перегонку, возгонку, экстракцию, диализ, осаждение, комплексообразование. Здесь мы остановимся на хроматографических методах, широко применяющихся как для разделения веществ, так и для их идентификации и количественного определения. Хроматографическое разделение основывается на различии таких свойств веществ, как летучесть, полярность, размер молекул, заряд и т.д. От них зависит распределение веществ между подвижной и неподвижной фазами, которые присутствуют в каждой хроматографической методике. Если подвижная фаза - газ, то метод называется газовой хроматографией (ГХ), если жидкость - жидкостной (ЖХ); если неподвижная фаза заполняет тонкую трубку или колонку, то это колоночная хроматография, а если она нанесена на пластину, то тонкослойная (ТСХ). Принципы разделения во всех случаях одинаковы, различаются только приборная реализация и методика. В тонкослойной хроматографии (рис. 12), например, образец наносят вблизи края пластинки с неподвижной фазой и погружают этот край в растворитель так, чтобы последний не доходил до места нанесения образца. Разделение проводят до тех пор, пока растворитель не дойдет до противоположного конца пластинки. Поскольку определяемые вещества движутся медленнее чистого растворителя, все они остаются на пластинке, но находятся на разном расстоянии от места нанесения образца. Скорость движения вещества характеризуется относительной разностью хода Rf :

В колоночной хроматографии (рис. 13) подвижная фаза непрерывно движется через неподвижную. Смесь, содержащую определяемые вещества, вводят с подвижной фазой в верхнюю часть колонки. Определяемые вещества вымываются из колонки потоком растворителя и проходят через детектор, который определяет их концентрацию. Скорость вымывания характеризуется параметром tr - временем, за которое концентрация вещества на выходе из колонки достигает максимума. Площадь пика на получающейся хроматограмме соответствует количеству вышедшего из колонки вещества.
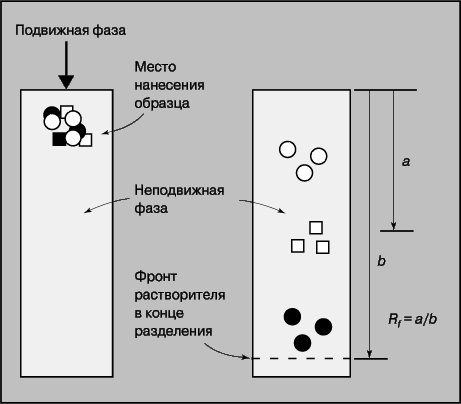
Рис. 12. ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ. 1 - подвижная фаза; 2 - место нанесения образца; 3 - неподвижная фаза; 4 - фронт растворителя в конце разделения.
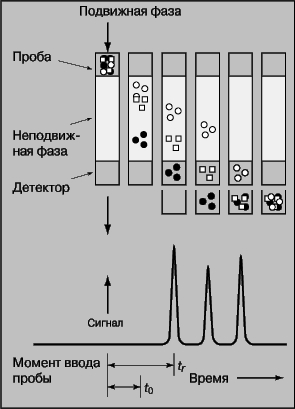
Рис. 13. КОЛОНОЧНАЯ ХРОМАТОГРАФИЯ. 1 - подвижная фаза; 2 - внесенный образец; 3 - неподвижная фаза; 4 - детектор.
Механизмы хроматографического разделения. Рассмотрим основные механизмы распределения веществ между подвижной и неподвижной фазами на примере колоночной хроматографии.
Адсорбционная хроматография. Неподвижная фаза представляет собой твердое вещество, на активных центрах которого адсорбируются молекулы определяемых веществ. Разделение может быть основано на различиях их полярностей: чем полярнее вещество, тем сильнее оно адсорбируется на неподвижной фазе и дольше задерживается на ней.
Распределительная хроматография. Неподвижная фаза представляет собой текучее вещество, нанесенное на твердый носитель или химически связанное с ним. Компоненты смеси, пропускаемой через колонку, разделяются вследствие разной растворимости в неподвижной фазе. Как в газовой, так и в жидкостной распределительной хроматографии используют неподвижные фазы с разной полярностью и другими химическими свойствами, от которых зависит растворимость определяемых веществ. Одна из разновидностей распределительной хроматографии - газожидкостная хроматография (ГЖХ) с жидкой неподвижной фазой и газовой подвижной. На рис. 14 представлена газожидкостная хроматограмма лимонного масла.
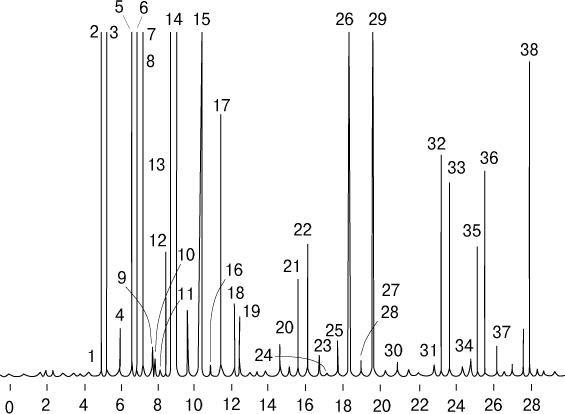
Рис. 14. ГАЗОЖИДКОСТНАЯ ХРОМАТОГРАФИЯ. Разделение лимонного масла. Пронумерованные пики отвечают различным компонентам лимонного масла, например, пик 8 - мирцен, 14 - лимонен, 20 - цитронеллаль, 25 - гераниол.
Вытеснительная хроматография. Неподвижная фаза представляет собой твердое пористое вещество. Крупные молекулы разделяемой смеси не проникают в поры и, не задерживаясь в неподвижной фазе, увлекаются растворителем. Молекулы среднего размера застревают в некоторых порах и на какое-то время остаются в неподвижной фазе, а мелкие молекулы проникают во все поры и перемещаются очень медленно. Так происходит разделение молекул по размерам, а следовательно, по молекулярной массе. Методом вытеснительной хроматографии можно разделять вещества с мол. массой от 100 до 100 000 000.
Ионообменная хроматография. Неподвижной фазой является ионит - твердое, практически нерастворимое в воде и органических растворителях вещество, содержащее ионогенные функциональные группы, способные обменивать свои ионы на ионы, присутствующие в подвижной фазе. Для разделения анионов (органических кислот, аминокислот или хлорид-, нитрат-, сульфат-ионов) используются аниониты, содержащие аминогруппу или четвертичный аммоний. В состав катионитов, применяемых для разделения катионов (аминокислот или ионов металлов), входят карбоновые или сульфокислоты.
Разделение оптически активных веществ. Многие оптически активные (хиральные) соединения (например, биологические молекулы, лекарственные вещества) обладают весьма ценными свойствами, но эти свойства часто бывают присущи только одному из изомеров. Разделить энантиомеры (зеркальные изомеры) при помощи традиционных хроматографических методов не удается, поскольку они обладают идентичными физическими и химическими свойствами, если не считать способности по-разному вращать плоскость поляризации света и взаимодействовать с другими хиральными соединениями. Последнее свойство и лежит в основе методов разделения энантиомеров. Один из подходов состоит в том, что проводят реакцию между смесью оптически активных определяемых веществ и каким-либо хиральным реагентом. Образующиеся продукты обладают разными физическими свойствами, и их разделяют обычными хроматографическими методами. В другом, более распространенном случае хиральное вещество используют в качестве неподвижной фазы для колоночной ЖХ. Энантиомеры по-разному взаимодействуют с ним и разделяются.
Тонкослойная хроматография (ТСХ). Неподвижная фаза - мелкодисперсный сорбент (обычно силикагель), нанесенный на стеклянную или металлическую пластину. На слой сорбента пипеткой наносят анализируемую смесь и пластину ставят торцом в растворитель. Под действием капиллярных сил растворитель поднимается по пластине, и смесь разделяется на компоненты. Флуоресцирующие вещества выявляют в УФ-свете, все остальные - с помощью специфических реагентов. ТСХ - простой, нетрудоемкий метод. На одной пластинке можно одновременно разделять несколько смесей, а для повышения эффективности проводить двумерную хроматографию.
СЕЛЕКТИВНЫЕ ОПРЕДЕЛЕНИЯ
Одна из основных задач аналитической химии заключается в достижении высокой селективности определений. В некоторых случаях селективность обеспечивается предварительным разделением исследуемых веществ, в других - совместным применением различных методов. Во многих современных системах применяются биологические объекты (ферменты, антитела и рецепторы) и специальные датчики. Датчики состоят из слоя химически активного вещества и физического преобразователя; их обычно используют для селективного измерения концентраций химических веществ. Кроме того, они позволяют проводить дистанционные и непрерывные измерения.
Ферментативные методы. Свойством ферментов, представляющим интерес для аналитической химии, является их способность специфически ускорять те или иные реакции. Ферментативные методы можно применять для анализа как равновесных, так и неравновесных систем, совмещать их с разными методами детектирования: спектрофотометрией, флуоресценцией, хемилюминесценцией, потенциометрией, амперометрией. Все чаще используются иммобилизованные ферменты. Нередко это повышает разрешающую способность метода, а кроме того, позволяет повторно использовать ферменты, применять их в проточных реакторах или биосенсорах. Ферменты включают в мембраны, полимерный гель с поперечными сшивками или адсорбируют на твердой подложке.
Иммунологические методы. Антитела - это вещества, которые вырабатываются в организме позвоночных в ответ на появление в нем антигенов и специфически связываются с этими антигенами. Специфичность связывания определяется структурным соответствием антигена и вырабатываемого антитела. В иммунологических определениях используются меченые антигены. Так, в радиоиммунологическом анализе (РИА) меткой служит радиоактивный изотоп, обычно 125I. В последнее время стали широко применяться флуоресцентные, хемилюминесцентные, электроактивные метки и ферменты. При помощи иммунологических методов анализируют лекарственные вещества, гормоны (такие, как хорионический гонадотропин, по которому определяют беременность), выявляют возбудителей инфекционных заболеваний.
Электрохимические датчики. Наиболее известный электрохимический датчик - это ионоселективный электрод. На принципе ионоселективности работают газовый потенциометрический и ферментный электроды. В них мембрана электрода покрыта слоем химического вещества, который отделен от анализируемого раствора (или газа) второй мембраной, проницаемой для определяемого вещества. Потенциометрический газовый электрод регистрирует изменение положения равновесия химической реакции, протекающей в слое вещества на мембране электрода. В этой реакции участвует газ, диффундирующий через наружную мембрану. Когда его количество меняется, положение равновесия реакции сдвигается, и этот сдвиг регистрируется электродом. В датчике CO2 используется водородный электрод, покрытый тонким слоем бикарбоната. CO2, проникая через наружную мембрану, сдвигает положение равновесия реакции CO2 + H2O HCO3- + H+, и водородный электрод измеряет концентрацию ионов водорода. В потенциометрических ферментных электродах мембрану электрода покрывают ферментом (например, уреазой в случае определения мочевины). Разработаны потенциометрические датчики для определения аминокислот, пенициллина и других антибиотиков. В качестве ферментсодержащего слоя можно использовать бактерии, интактные растительные и животные ткани. В амперометрических ферментных электродах чаще всего ферментом является оксидаза и регистрируется либо расходование кислорода, либо образование пероксида водорода. Для контроля за содержанием глюкозы в биологических жидкостях применяются амперометрические датчики на основе глюкозооксидазы.
Оптические датчики. В таких датчиках специфический реагент наносят на торец оптического волокна - световода. По световоду направляют луч света и регистрируют свет, пришедший от торца с нанесенным образцом. Особенно много датчиков разработано для оптического измерения pH. Все они содержат иммобилизованный реагент, который может существовать в двух или более кислотно-основных формах. Если у этих форм разные спектры поглощения или флуоресценции, то, проводя измерения при разных длинах волн, можно определить их концентрацию и рассчитать pH. В отличие от стеклянного электрода, измеряющего pH в диапазоне от 1 до 14, у оптических датчиков динамический диапазон регистрируемых значений pH охватывает 1-2 единицы по обе стороны от pKa индикатора. В датчиках ионов металлов (Al3+, Mg2+, Zn2+, Cd2+) используют лиганды, которые начинают сильно флуоресцировать при связывании с этими ионами. Датчики кислорода основаны на кислородном подавлении иммобилизованного флуорофора. Это равновесное определение, менее восприимчивое к колебаниям температуры и скорости потока, чем амперометрические датчики кислорода. Разработаны биосенсоры, основанные на принципах иммунологического анализа. На торец оптических волокон таких датчиков наносят антитела и флуоресцентно меченные антигены.
Датчики массы. На чувствительный к изменению массы преобразователь (например, кварцевый пьезоэлектрический осциллятор) наносят селективный адсорбент. Определяемое вещество осаждается на нем, и датчик регистрирует изменение массы. Подобные датчики применяются для определения газообразных и летучих веществ, таких, как CO, CO2 и SO2, ароматических и алифатических углеводородов и пестицидов.
ЛИТЕРАТУРА
Крешков А. П. Основы аналитической химии, тт. 1-3. М., 1977 Слейбо У., Пергона Т. Общая химия. М., 1979 Карапетьянц М. Х., Дракин С.И. Общая химия. М., 1981 Глинка Н. Л. Общая химия. Л., 1988
Энциклопедия Кольера. — Открытое общество. 2000.
Полезное
Смотреть что такое "ХИМИЯ АНАЛИТИЧЕСКАЯ" в других словарях:
Химия высокомолекулярных соединений — Химия полимеров один из перспективных и успешно развивающихся разделов химической науки. Делится на разделы: физическая химия полимеров, структурная и т. д. Благодаря успешному развитию химии полимеров создаются новые материалы, нашедшие… … Википедия
ХИМИЯ — (возможно от греч. Chemia Хемия, одно из древнейших названий Египта), наука, изучающая превращения веществ, сопровождающиеся изменением их состава и (или) строения. Химические процессы (получение металлов из руд, крашение тканей, выделка кожи и… … Большой Энциклопедический словарь
химия — и; ж. [от араб. alchimia (ал)химия] 1. Научная дисциплина (область естествознания), изучающая вещества, их состав, строение, свойства и взаимные превращения. Прикладная х. Неорганическая х. Органическая х. Теоретическая х. Аналитическая х.… … Энциклопедический словарь
ХИМИЯ. РАЗДЕЛЫ — Химию довольно произвольно делят на несколько разделов, которые нельзя четко отграничить ни от других областей химии, ни от других наук (физики, геологии, биологии). Неорганическая химия занимается изучением химической природы элементов и их… … Энциклопедия Кольера
химия — сущ., ж., употр. сравн. часто Морфология: (нет) чего? химии, чему? химии, (вижу) что? химию, чем? химией, о чём? о химии 1. Химией называется область естествознания, которая изучает вещества, их превращения и способы управления этими… … Толковый словарь Дмитриева
Химия — (гр. Хемия, одно из древнейших названий Египта) наука, изучающая превращения веществ, сопровождающиеся изменением их состава и (или) строения. Хим. процессы использовались человечеством уже на заре его культурной жизни. В 3 4 вв. зародилась… … Концепции современного естествознания. Словарь основных терминов
ХИМИЯ — (возможно, от греч. Chemia Хемия, одно из древнейших назв. Египта), наука, изучающая превращения в в, сопровождающиеся изменением их состава и (или) строения. Хим. процессы (получение металлов из руд, крашение тканей, выделка кожи и др.)… … Естествознание. Энциклопедический словарь
Химия почв — Химия почв это раздел почвоведения, изучающий химические основы почвообразования и плодородия почв. Основой для решения этих вопросов служит исследование состава, свойств почв и протекающих в почвах процессов на ионно молекулярном и… … Википедия
ХИМИЯ — ХИМИЯ, наука о веществах, их превращениях, взаимодействии и о происходящих при этом явлениях. Выяснением основных понятий, к рыми оперирует X., как напр, атом, молекула, элемент, простое тело, реакция и др., учением о молекулярных, атомных и… … Большая медицинская энциклопедия
АНАЛИТИЧЕСКАЯ ХИМИЯ — рассматривает принципы и методы определения химического состава вещества. Включает качественный анализ и количественный анализ. Аналитическая химия возникла наряду с неорганической химией раньше других химических наук (до кон. 18 в. химия… … Большой Энциклопедический словарь
 18+© Академик, 2000-2024
18+© Академик, 2000-2024- Обратная связь: Техподдержка, Реклама на сайте
Экспорт словарей на сайты, сделанные на PHP, Joomla, Drupal, WordPress, MODx.