- Углеводороды ароматические
-
получили свое название оттого, что очень часто их производные обладают приятным запахом и встречаются в различных смолах, эфирных маслах и пр. Главным продуктом для их получения служит каменноугольная смола, образующаяся в довольно значительном количестве при производстве светильного газа. У. ароматические представляют очень обширный и очень важный класс У. вообще: их производные приобретают все большее и большее значение в различных отраслях техники, из них готовят многие медицинские препараты, органические краски, ароматические жидкости, дезинфекционные средства и пр. и пр. В научном отношении эти соединения доставляют также весьма много интереса, так как по многим своим свойствам они резко отличаются от других органических соединений и эти особенности нужно приписать непременному присутствию в них специальной группировки из 6 атомов углерода, называемой бензольным кольцом, которая может в частице аромат. У. повторяться и несколько раз. Это же бензольное кольцо указывает на то, что простейшим аром. У. может быть только углеводород, содержащий не менее 6 атомов углерода, и действительно, подобно тому как метан СН4 (см.) начинает собой ряд жирных У., которые все могут быть от него произведены, так и бензол C6H6 есть простейший родоначальник всех аром. У., не содержащий в своей молекуле никаких других группировок, кроме бензольного кольца, а так как выше было упомянуто, что это кольцо и обусловливает специфичность свойств аром. У., то, очевидно, в бензоле она выступает особенно резко, так как не маскируется здесь свойствами других атомных группировок, и потому неудивительно, что бензол с момента его открытия постоянно служил предметом многочисленных научных исследований, которые, с одной стороны, имели целью более точное определение характера ароматических соединений вообще, а с другой, были направлены к объяснению строения бензольного кольца. Несмотря на то, что бензол был открыт еще в 1825 г. Фарадеем, более или менее научно обоснованная теория аром. У. и их производных была предложена Кекуле только в 1865 г. Этот великий химик сумел обобщить отрывочные данные относительно строения аром. У. и смело представил теорию бензольных, или ароматических, соединений, сравнительно весьма мало обоснованную, но которая впоследствии, по крайней мере в своей основе, подтвердилась блестящим образом. Теория Кекуле заключается в следующих 3-х положениях: 1) все ароматические соединения происходят от ядра, содержащего 6 атомов углерода, и их простейший представитель есть бензол C6H6. Через замену в нем одного или нескольких атомов водорода другими атомами или атомными группами (как их называет Кекуле, боковыми цепями) и получаются ароматические соединения, которые все без исключения удерживают основные черты характера бензола и потому могут быть рассматриваемы как его производные. 2) Бензол имеет симметрическое строение, и в нем каждый атом углерода связан с одним атомом водорода, образуя карбинную группу ≡СН. Это заключение выведено на основании того, что не известно изомерных (см.) монопроизводных бензола и явления изомерии появляются только при ди- и полипроизводных его. 3) Принимая во внимание четырехатомность углерода, на основании положения 2-го бензол может обладать только следующей структурной формулой:
 в которой двойные связи между карбинными группами чередуются простыми [Как показано будет далее, это 3-e положение в настоящее время принимается далеко не всеми учеными.]. Ясно, что центр тяжести всей теории Кекуле лежал во втором положении и его требовалось доказать самым строгим образом, что и было выполнено весьма изящно Ладенбургом, Вроблевским, Гюбнером и Петерманном. Прежде всего, Ладенбург в 1874 г. показал, что в бензоле 4 атома водорода совершенно равнозначны между собой. Это заключение им было сделано на основании следующих данных опыта: в феноле, т. е. бензоле, в котором один водород замещен оксигруппой ОН, можно эту группу заместить карбоксилом СО2Н и получить бензойную кислоту С6Н5—СО2Н, в которой, следовательно, группа CO2H будет связана с тем же углеродным атомом, положим 3-м, как и группа ОН в первоначально взятом феноле. Далее, известны были три монооксибензойных кислоты, т. е. три таких бензойных кислоты, в которых еще один из водородов бензольного ядра был замещен оксигруппой, следовательно, они имели формулу C6H4(OH)—CO2H; но так как эти кислоты различны и, кроме того, одна из них получается из бензойной кислоты, а другие две могут быть переведены в эту последнюю, то, следовательно, в этих оксибензойных кислотах карбоксильная группа также связана с 1-м углеродом, а их оксигруппы стоят при различных углеродных атомах, положим, при 2, 3 и 4. Эти оксибензойные кислоты при прокаливании с известью дали Ладенбургу один и тот же фенол C6H5OH, идентичный с тем, который им был взят для получения бензойной кислоты; но ведь в оксибензойных кислотах ОН-группы стояли при 2, 3 и 4 углеродах, и так как при замещении водородных атомов при углеродах 1, 2, 3 и 4 группами ОН получается всегда один и тот же фенол, то ясно, что эти водородные атомы равнозначны между собой. Несколько раньше, гораздо более сложным путем Ладенбург доказал, что в бензоле всегда имеются две пары атомов водорода, симметричных по отношению к пятому атому водорода, однако это доказательство хотя и увеличивало степень равнозначности некоторых водородных атомов бензола, однако еще не доказывало ее вполне. Гораздо проще этого сделали Гюбнер и Петерманн для одной пары и позднее Вроблевский — для другой. Первые ученые, исходя из бромбензойной кисл. С6Н4Br—CO2Н, в которой, положим, были замещены карбоксилом и бромом водороды при углеродах 1 и 3, получили нитрацией две нитробензойных кислоты, в которых, очевидно, СО2Н и Br стояли при прежних углеродах 1 и 3, группы же нитро могли находиться при любом из остальных углеродов, положим, в первой кисл. — при 2, а во второй — при 6. Эти нитрокислоты при восстановлении дали одну и ту же амидобензойную кислоту C6H4(NH2)—CO2H, в которой, очевидно, группа СО2H занимала прежнее место 1, а группа же NH2 в случае первой нитрокислоты должна была занимать место 2, а в случае второй — место 6; так как эти кислоты получились идентичными, то, следовательно, это могло случиться только тогда, когда водородные атомы при углеродах 2 и 6 симметричны по отношению к углероду 1. Далее, Вроблевский, бромируя ацетотолуидин C6H4(CH3)—NH—C2H3O, в котором были, положим, замещены водороды ядра 1 и 2 группами СН3 и NHC2H3O, получил бромацетотолуидин С6Н3(СН3)(NH—C2Н3О)Br, т. е. заместил еще один водород, положим, 3, затем он полученный продукт подверг двоякого рода обработке; с одной стороны, ацетаминная группа была замещена водородом и полученный бромтолуол СбН4—СН3Br при окислении дал бромбензойную кислоту, тождественную с той, из которой исходили при своих работах Гюбнер и Петерманн и в которой мы принимали замещенными 1 и 3 атомы водорода, след., и бромтолуол Вроблевского имеет бензольное кольцо с замещенными водородами при 1 и 3 углероде. С другой стороны, Вроблевский в полученном им ацетобромтолуидине заместил еще один водород, положим — 4-й, группой NO2, далее в полученном теле заместил ацетаминогруппу водородом и при последующем восстановлении получил толуидин C6H4(CH3)NH2, в котором. очевидно, СН3 занимал прежнее место 1-е, a NH2 — место группы NO2, т. е. 4-е; этот толуидин он окончательно превратил в бромтолуол С6Н4СН3Br заменой группы NH2 бромом; таким образом, в этом бромтолуоле опять-таки были замещенными места 1 и 4; по исследовании же полученного вещества оказалось, что оно совершенно тождественно с бромтолуолом, приготовленным по первому способу, и отсюда ясно, что места 3 и 4 симметричны по отношению к 1. Итак, из работ Гюбнера и Петерманна вытекает симметричность мест 2 и 6 по отношению к месту 1, а из работ Вроблевского — мест 3 и 4. Следовательно, в бензоле всегда имеются две пары атомов водорода, расположенных симметрично по отношению к пятому атому. Наконец, бромбензойная кислота 1, 3 легко получается из бензойной, а потому в ней карбоксильная группа замещает тот же атом водорода, как в бензойной кислоте Ладенбурга, т. е. при 1-м углероде; этот же атом совершенно равнозначен с четырьмя другими, и следовательно, в бензоле не только для одного, но и для четырех атомов углерода должны иметься по 2 пары симметрических атомов, и это следствие уже необходимо влечет за собой заключение, что все шесть углеродных атомов в бензоле равнозначны, т. е. что бензол состоит из шести групп СН, одинаково связанных между собой. Таким образом было строго доказано второе положение Кекуле, что же касается вопроса о том, каким образом в бензоле связаны между собою группы СН, то он до сих пор остается открытым, несмотря на многочисленные попытки решить его. Но если строение бензола еще не вполне доказано, тем не менее вполне установленная идентичность в нем карбинных групп (СН) и кольчатость их расположения уже вносит много в химию аром. У.; именно эти положения теоретически предсказывают возможность существования только одного видоизменения монопроизводных бензола, трех двузамещенных производных его, 3-х тризамещенных, 3-х четырехзамещенных и одного пяти- и шестизамещенного производного. Этот вывод становится очевидным, если представить собе бензольное кольцо в виде правильного шестиугольника, в вершинах которого будут находиться углеродные атомы карбинных групп; если затем перенумеровать эти углероды, как показано на чертеже, то очевидно, что у какого бы из углеродов 1—б ни замещать водород, положим, какой-нибудь группой а, всегда получится одно и то же тело С6H5а; замещая еще один водород такой же группой а, на основании приведенного чертежа возможно предположить существование 3-х изомеров формулы C6H4a, в которых группы a будут находиться при углеродах 1 и 2, 1 и 3, 1 и 4, и очевидно, что большего числа изомеров в этом случае представить нельзя, так как возможные еще с первого взгляда изомеры 1 и 5, 1 и 6, конечно, будут тождественны с 1, 2 и 1, 3.
в которой двойные связи между карбинными группами чередуются простыми [Как показано будет далее, это 3-e положение в настоящее время принимается далеко не всеми учеными.]. Ясно, что центр тяжести всей теории Кекуле лежал во втором положении и его требовалось доказать самым строгим образом, что и было выполнено весьма изящно Ладенбургом, Вроблевским, Гюбнером и Петерманном. Прежде всего, Ладенбург в 1874 г. показал, что в бензоле 4 атома водорода совершенно равнозначны между собой. Это заключение им было сделано на основании следующих данных опыта: в феноле, т. е. бензоле, в котором один водород замещен оксигруппой ОН, можно эту группу заместить карбоксилом СО2Н и получить бензойную кислоту С6Н5—СО2Н, в которой, следовательно, группа CO2H будет связана с тем же углеродным атомом, положим 3-м, как и группа ОН в первоначально взятом феноле. Далее, известны были три монооксибензойных кислоты, т. е. три таких бензойных кислоты, в которых еще один из водородов бензольного ядра был замещен оксигруппой, следовательно, они имели формулу C6H4(OH)—CO2H; но так как эти кислоты различны и, кроме того, одна из них получается из бензойной кислоты, а другие две могут быть переведены в эту последнюю, то, следовательно, в этих оксибензойных кислотах карбоксильная группа также связана с 1-м углеродом, а их оксигруппы стоят при различных углеродных атомах, положим, при 2, 3 и 4. Эти оксибензойные кислоты при прокаливании с известью дали Ладенбургу один и тот же фенол C6H5OH, идентичный с тем, который им был взят для получения бензойной кислоты; но ведь в оксибензойных кислотах ОН-группы стояли при 2, 3 и 4 углеродах, и так как при замещении водородных атомов при углеродах 1, 2, 3 и 4 группами ОН получается всегда один и тот же фенол, то ясно, что эти водородные атомы равнозначны между собой. Несколько раньше, гораздо более сложным путем Ладенбург доказал, что в бензоле всегда имеются две пары атомов водорода, симметричных по отношению к пятому атому водорода, однако это доказательство хотя и увеличивало степень равнозначности некоторых водородных атомов бензола, однако еще не доказывало ее вполне. Гораздо проще этого сделали Гюбнер и Петерманн для одной пары и позднее Вроблевский — для другой. Первые ученые, исходя из бромбензойной кисл. С6Н4Br—CO2Н, в которой, положим, были замещены карбоксилом и бромом водороды при углеродах 1 и 3, получили нитрацией две нитробензойных кислоты, в которых, очевидно, СО2Н и Br стояли при прежних углеродах 1 и 3, группы же нитро могли находиться при любом из остальных углеродов, положим, в первой кисл. — при 2, а во второй — при 6. Эти нитрокислоты при восстановлении дали одну и ту же амидобензойную кислоту C6H4(NH2)—CO2H, в которой, очевидно, группа СО2H занимала прежнее место 1, а группа же NH2 в случае первой нитрокислоты должна была занимать место 2, а в случае второй — место 6; так как эти кислоты получились идентичными, то, следовательно, это могло случиться только тогда, когда водородные атомы при углеродах 2 и 6 симметричны по отношению к углероду 1. Далее, Вроблевский, бромируя ацетотолуидин C6H4(CH3)—NH—C2H3O, в котором были, положим, замещены водороды ядра 1 и 2 группами СН3 и NHC2H3O, получил бромацетотолуидин С6Н3(СН3)(NH—C2Н3О)Br, т. е. заместил еще один водород, положим, 3, затем он полученный продукт подверг двоякого рода обработке; с одной стороны, ацетаминная группа была замещена водородом и полученный бромтолуол СбН4—СН3Br при окислении дал бромбензойную кислоту, тождественную с той, из которой исходили при своих работах Гюбнер и Петерманн и в которой мы принимали замещенными 1 и 3 атомы водорода, след., и бромтолуол Вроблевского имеет бензольное кольцо с замещенными водородами при 1 и 3 углероде. С другой стороны, Вроблевский в полученном им ацетобромтолуидине заместил еще один водород, положим — 4-й, группой NO2, далее в полученном теле заместил ацетаминогруппу водородом и при последующем восстановлении получил толуидин C6H4(CH3)NH2, в котором. очевидно, СН3 занимал прежнее место 1-е, a NH2 — место группы NO2, т. е. 4-е; этот толуидин он окончательно превратил в бромтолуол С6Н4СН3Br заменой группы NH2 бромом; таким образом, в этом бромтолуоле опять-таки были замещенными места 1 и 4; по исследовании же полученного вещества оказалось, что оно совершенно тождественно с бромтолуолом, приготовленным по первому способу, и отсюда ясно, что места 3 и 4 симметричны по отношению к 1. Итак, из работ Гюбнера и Петерманна вытекает симметричность мест 2 и 6 по отношению к месту 1, а из работ Вроблевского — мест 3 и 4. Следовательно, в бензоле всегда имеются две пары атомов водорода, расположенных симметрично по отношению к пятому атому. Наконец, бромбензойная кислота 1, 3 легко получается из бензойной, а потому в ней карбоксильная группа замещает тот же атом водорода, как в бензойной кислоте Ладенбурга, т. е. при 1-м углероде; этот же атом совершенно равнозначен с четырьмя другими, и следовательно, в бензоле не только для одного, но и для четырех атомов углерода должны иметься по 2 пары симметрических атомов, и это следствие уже необходимо влечет за собой заключение, что все шесть углеродных атомов в бензоле равнозначны, т. е. что бензол состоит из шести групп СН, одинаково связанных между собой. Таким образом было строго доказано второе положение Кекуле, что же касается вопроса о том, каким образом в бензоле связаны между собою группы СН, то он до сих пор остается открытым, несмотря на многочисленные попытки решить его. Но если строение бензола еще не вполне доказано, тем не менее вполне установленная идентичность в нем карбинных групп (СН) и кольчатость их расположения уже вносит много в химию аром. У.; именно эти положения теоретически предсказывают возможность существования только одного видоизменения монопроизводных бензола, трех двузамещенных производных его, 3-х тризамещенных, 3-х четырехзамещенных и одного пяти- и шестизамещенного производного. Этот вывод становится очевидным, если представить собе бензольное кольцо в виде правильного шестиугольника, в вершинах которого будут находиться углеродные атомы карбинных групп; если затем перенумеровать эти углероды, как показано на чертеже, то очевидно, что у какого бы из углеродов 1—б ни замещать водород, положим, какой-нибудь группой а, всегда получится одно и то же тело С6H5а; замещая еще один водород такой же группой а, на основании приведенного чертежа возможно предположить существование 3-х изомеров формулы C6H4a, в которых группы a будут находиться при углеродах 1 и 2, 1 и 3, 1 и 4, и очевидно, что большего числа изомеров в этом случае представить нельзя, так как возможные еще с первого взгляда изомеры 1 и 5, 1 и 6, конечно, будут тождественны с 1, 2 и 1, 3. Для отличия друг от друга этих изомеров обыкновенно перед их названием ставят цифры, указывающие, у каких углеродов находятся боковые цепи, или прибавляют к названию приставки орто- (см.), мета- (см.) и пара- (см.). Напр., говорят 1,2-дихлорбензол, или ортодихлорбензол, 1,4-динитробензол, или парадинитробензол, и т. д. При замещении водородов бензольного кольца тремя группами а получаются изомеры:1 2, 3 или v. или рядовой.1 2 4 " as. " асимметрический.1 3 5 " s. " симметрический.Для четырехзамещенных C6H2a имеем: 1, 2, 3, 4, или v, l, 2, 4, 5, или s, и 1, 2, 3, 5, или as. Очевидно, что это число изомеров значительно увеличивается, если замещающие группы будут не одинаковы между собой, и, напр., производных типа С6Н3Х2У может быть не три, а 6: (1, 2, 3), (l, 2, 4), (1, 2, 5), (1, 2, 6), (1, 3, 4), (1, 3, 5), причем в этом обозначении принято, что группа Y всегда занимает место у углерода 1. Так как указанные факты вытекают как следствие из не подлежащего сомнению положения 2-го теории Кекуле, то ничего нет удивительного, что они подтверждаются на практике самым блестящим образом. Каким же образом возможно подтвердить их практически? Другими словами, как доказать, что данный изомер имеет в этом случае то, а не другое строение? Обыкновенно для этой цели стараются превратить изучаемый изомер в такое соединение, строение которого строго определено. Для аром. У. особенное значение в этом случае имеют бензолдикарбоновые, или фталевые, кислоты, потому что при окислении боковых углеводородных цепей ароматических соединений они всегда превращаются в конце концов в группы СО2Н, и, след., аром. У. при энергичном окислении вообще всегда дают карбоновые кисл., карбоксильные группы которых связаны с теми углеродами ядра, с какими были связаны углеводородные боковые цепи. Следовательно, аром. У. с одной боковой цепью дает при окислении C6H5CO2H — бензойную кисл., с 2-мя боковыми цепями дает С6H4(СО2Н)2 — одну из фталевых кислот и т. д. Строение же трех возможных фталевых кислот:
Для отличия друг от друга этих изомеров обыкновенно перед их названием ставят цифры, указывающие, у каких углеродов находятся боковые цепи, или прибавляют к названию приставки орто- (см.), мета- (см.) и пара- (см.). Напр., говорят 1,2-дихлорбензол, или ортодихлорбензол, 1,4-динитробензол, или парадинитробензол, и т. д. При замещении водородов бензольного кольца тремя группами а получаются изомеры:1 2, 3 или v. или рядовой.1 2 4 " as. " асимметрический.1 3 5 " s. " симметрический.Для четырехзамещенных C6H2a имеем: 1, 2, 3, 4, или v, l, 2, 4, 5, или s, и 1, 2, 3, 5, или as. Очевидно, что это число изомеров значительно увеличивается, если замещающие группы будут не одинаковы между собой, и, напр., производных типа С6Н3Х2У может быть не три, а 6: (1, 2, 3), (l, 2, 4), (1, 2, 5), (1, 2, 6), (1, 3, 4), (1, 3, 5), причем в этом обозначении принято, что группа Y всегда занимает место у углерода 1. Так как указанные факты вытекают как следствие из не подлежащего сомнению положения 2-го теории Кекуле, то ничего нет удивительного, что они подтверждаются на практике самым блестящим образом. Каким же образом возможно подтвердить их практически? Другими словами, как доказать, что данный изомер имеет в этом случае то, а не другое строение? Обыкновенно для этой цели стараются превратить изучаемый изомер в такое соединение, строение которого строго определено. Для аром. У. особенное значение в этом случае имеют бензолдикарбоновые, или фталевые, кислоты, потому что при окислении боковых углеводородных цепей ароматических соединений они всегда превращаются в конце концов в группы СО2Н, и, след., аром. У. при энергичном окислении вообще всегда дают карбоновые кисл., карбоксильные группы которых связаны с теми углеродами ядра, с какими были связаны углеводородные боковые цепи. Следовательно, аром. У. с одной боковой цепью дает при окислении C6H5CO2H — бензойную кисл., с 2-мя боковыми цепями дает С6H4(СО2Н)2 — одну из фталевых кислот и т. д. Строение же трех возможных фталевых кислот: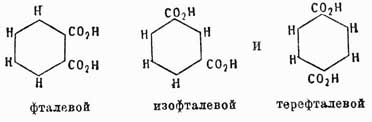 было доказано следующим образом. Фталевая кисл. получается при окислении аром. У. нафталина С10Н8; с другой стороны, нитруя предварительно нафталин, мы получаем нитронафталин С10Н7—NO3, дающий при окислении нитробензолдикарбоновую кислоту, которая по восстановлении группы NO2 в амидогруппу и замене последней водородом превращается опять-таки во фталевую кислоту. Отсюда мы можем заключить, что нафталин содержит одно бензольное ядро, связанное с двумя углеводородными боковыми цепями. Теперь далее, если до окисления восстановить нитронафталин в амидопроизводное и затем это последнее окислить, то снова получается не амидофталевая кислота, как в первом случае, а просто фталевая, поэтому нужно признать, что во втором случае сгорело при окислении первое бензольное ядро, в котором находилась NH2, а следов., и NO2-группа; но образовавшаяся фталевая кислота указывает, что в нафталине имеется еще одно бензольное кольцо, однако эмпирическая формула этого У. C10H8 не допускает этого; затрудение возможно разрешить, предположив только, что нафталин состоит из двух бензольных колец, сочлененных между собой так, как это показано на чертеже:
было доказано следующим образом. Фталевая кисл. получается при окислении аром. У. нафталина С10Н8; с другой стороны, нитруя предварительно нафталин, мы получаем нитронафталин С10Н7—NO3, дающий при окислении нитробензолдикарбоновую кислоту, которая по восстановлении группы NO2 в амидогруппу и замене последней водородом превращается опять-таки во фталевую кислоту. Отсюда мы можем заключить, что нафталин содержит одно бензольное ядро, связанное с двумя углеводородными боковыми цепями. Теперь далее, если до окисления восстановить нитронафталин в амидопроизводное и затем это последнее окислить, то снова получается не амидофталевая кислота, как в первом случае, а просто фталевая, поэтому нужно признать, что во втором случае сгорело при окислении первое бензольное ядро, в котором находилась NH2, а следов., и NO2-группа; но образовавшаяся фталевая кислота указывает, что в нафталине имеется еще одно бензольное кольцо, однако эмпирическая формула этого У. C10H8 не допускает этого; затрудение возможно разрешить, предположив только, что нафталин состоит из двух бензольных колец, сочлененных между собой так, как это показано на чертеже: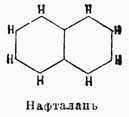 и тогда все вышеуказанные переходы легко объясняются следующей схемой, которая и устанавливает строение фталевой кислоты:
и тогда все вышеуказанные переходы легко объясняются следующей схемой, которая и устанавливает строение фталевой кислоты: Изофталевая кислота имеет строение метадикарбоновой кислоты, потому что она получается из мезитилена, который есть симметрический триметилбензол С6Н3(СН3)3, и схема ее получения следующая:
Изофталевая кислота имеет строение метадикарбоновой кислоты, потому что она получается из мезитилена, который есть симметрический триметилбензол С6Н3(СН3)3, и схема ее получения следующая: Симметричность строения мезитилена была весьма строго доказана Ладенбургом. Итак, следовательно, из трех фталевых кислот строение двух строго доказано, а для третьей — терефталевой кислоты — остается, значит, строение единственно возможного еще изомера 1, 4. Для доказательства строения три- и тетразамещенных бензолов можно пользоваться, исходя из фталевых кислот, методом, который Кёрнер применил для доказательства строения дибром- и трибромбензолов. Метод этот заключается в следующем: если мы имеем три изомера типа C6H4X2, то, переводя их в соединения типа С6Н3Х3, можно предполагать, что:
Симметричность строения мезитилена была весьма строго доказана Ладенбургом. Итак, следовательно, из трех фталевых кислот строение двух строго доказано, а для третьей — терефталевой кислоты — остается, значит, строение единственно возможного еще изомера 1, 4. Для доказательства строения три- и тетразамещенных бензолов можно пользоваться, исходя из фталевых кислот, методом, который Кёрнер применил для доказательства строения дибром- и трибромбензолов. Метод этот заключается в следующем: если мы имеем три изомера типа C6H4X2, то, переводя их в соединения типа С6Н3Х3, можно предполагать, что: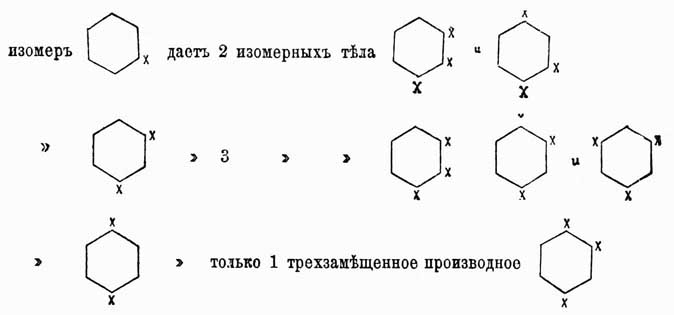 Из рассмотрения этой таблицы ясно, что изомер 1, 3, 4 образуется из всех трех двузамещенных, изомер 1, 2, 3 только из двух и изомер 1, 3, 5 из одного двузамещенного бензола; можно вывести и обратное заключение: исходя из трехзамещенного бензола 1, 3,4, можно получить все три двузамещенных и т. д. Комбинируя так или иначе эти данные, легко прийти к строгому определению строения трех- и вообще полизамещенных бензолов. Благодаря разработке вышеуказанных методов строение различных ароматических соединений в громадном большинстве случаев стало ясно, и явилась возможность приступить к изучению свойств различных изомеров в зависимости от расположения боковых групп, и здесь в первый раз появились факты, противоречащие формуле строения бензола, данной Кекуле (см. выше). По этой формуле бензол является У. непредельным, однако, на самом деле его непредельный характер очень слабо развит; правда, бензол способен водородом в момент выделения восстановляться, присоединяя 6 атомов водорода и давая гексаметилен:
Из рассмотрения этой таблицы ясно, что изомер 1, 3, 4 образуется из всех трех двузамещенных, изомер 1, 2, 3 только из двух и изомер 1, 3, 5 из одного двузамещенного бензола; можно вывести и обратное заключение: исходя из трехзамещенного бензола 1, 3,4, можно получить все три двузамещенных и т. д. Комбинируя так или иначе эти данные, легко прийти к строгому определению строения трех- и вообще полизамещенных бензолов. Благодаря разработке вышеуказанных методов строение различных ароматических соединений в громадном большинстве случаев стало ясно, и явилась возможность приступить к изучению свойств различных изомеров в зависимости от расположения боковых групп, и здесь в первый раз появились факты, противоречащие формуле строения бензола, данной Кекуле (см. выше). По этой формуле бензол является У. непредельным, однако, на самом деле его непредельный характер очень слабо развит; правда, бензол способен водородом в момент выделения восстановляться, присоединяя 6 атомов водорода и давая гексаметилен: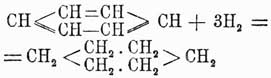 способен также на солнечном свету присоединять 6 атомов галоида; но он не присоединяет галоидоводородных кислот. Также в отличие от жирных углеводородов его галоидопроизводные, в которых галоиды не присоединены к бензолу, а замещают водороды его карбинных групп, совершенно почти не обмыливаются щелочами. Все эти факты заставляли несколько усомниться в правильности формулы Кекуле; при изучении же двузамещенных производных бензола эта формула уже подверглась сильному сомнению, и ей начали противополагать многие другие. В самом деле, на основании формулы Кекуле нужно полагать, что существуют два ортоизомера: один из них образуется замещением водородов при соседних углеродах, связанных между собой двойной, а другой замещением водородов при углеродах, связанных простой связью. Однако этого в действительности никогда не наблюдается, и все попытки выделить подобные изомеры не имели успеха. Для объяснения этого противоречия Кекуле предложил теорию колеблющихся связей (oscillirenden Bindungen), по которой двойные связи в бензольном кольце не имеют постоянного положения, и, напр., двойная связь при углероде 1-м может связывать с ним то 2-й углерод, то 6-й, а потому очень естественно, что при таком условии не может существовать и двух ортоизомеров. Однако такая теория является совершенно недоказанной, тем более, что несуществование двух ортоизомеров можно объяснить и другим образом, именно: мы знаем, что двойная связь в жирных соединениях характеризует собой некоторую неустойчивость равновесия данного химического соединения и, стремясь постоянно прийти к более устойчивой форме, она обусловливает собой в соединении более развитую способность вступать в различные реакции, а потому нет ничего удивительного, что в бензоле при образовании двузамещенных соединений эти последние всегда отвечают формуле, в которой замещены водороды при углеродах, связанных двойной связью. Тем не менее, каково бы ни было объяснение этого противоречия формулы Кекуле с действительностью, оно все-таки имеет известные натяжки и допущение этой формулой двух ортоизомеров является ее слабой стороной. Далее, сравнительное изучение орто-, пара- и метаизомеров показало, что первые два всегда очень близки по свойствам друг к другу и довольно сильно отличаются от последнего. Этот факт опять-таки говорит против формулы Кекуле, на основании которой скорее можно было бы предполагать постепенное изменение свойств изомеров по мере удаления друг от друга боковых цепей, т. е., другими словами, нужно было бы ожидать, что свойства метаизомера будут средними между свойствами орто- и парасоединений. Это противоречие уже совершенно не объяснялось теорией и вызвало несколько новых формул строения бензола, основанием для которых было 2-ое положение Кекуле и близость свойств орто- и параизомеров. Таковыми являются формулы: Клауса
способен также на солнечном свету присоединять 6 атомов галоида; но он не присоединяет галоидоводородных кислот. Также в отличие от жирных углеводородов его галоидопроизводные, в которых галоиды не присоединены к бензолу, а замещают водороды его карбинных групп, совершенно почти не обмыливаются щелочами. Все эти факты заставляли несколько усомниться в правильности формулы Кекуле; при изучении же двузамещенных производных бензола эта формула уже подверглась сильному сомнению, и ей начали противополагать многие другие. В самом деле, на основании формулы Кекуле нужно полагать, что существуют два ортоизомера: один из них образуется замещением водородов при соседних углеродах, связанных между собой двойной, а другой замещением водородов при углеродах, связанных простой связью. Однако этого в действительности никогда не наблюдается, и все попытки выделить подобные изомеры не имели успеха. Для объяснения этого противоречия Кекуле предложил теорию колеблющихся связей (oscillirenden Bindungen), по которой двойные связи в бензольном кольце не имеют постоянного положения, и, напр., двойная связь при углероде 1-м может связывать с ним то 2-й углерод, то 6-й, а потому очень естественно, что при таком условии не может существовать и двух ортоизомеров. Однако такая теория является совершенно недоказанной, тем более, что несуществование двух ортоизомеров можно объяснить и другим образом, именно: мы знаем, что двойная связь в жирных соединениях характеризует собой некоторую неустойчивость равновесия данного химического соединения и, стремясь постоянно прийти к более устойчивой форме, она обусловливает собой в соединении более развитую способность вступать в различные реакции, а потому нет ничего удивительного, что в бензоле при образовании двузамещенных соединений эти последние всегда отвечают формуле, в которой замещены водороды при углеродах, связанных двойной связью. Тем не менее, каково бы ни было объяснение этого противоречия формулы Кекуле с действительностью, оно все-таки имеет известные натяжки и допущение этой формулой двух ортоизомеров является ее слабой стороной. Далее, сравнительное изучение орто-, пара- и метаизомеров показало, что первые два всегда очень близки по свойствам друг к другу и довольно сильно отличаются от последнего. Этот факт опять-таки говорит против формулы Кекуле, на основании которой скорее можно было бы предполагать постепенное изменение свойств изомеров по мере удаления друг от друга боковых цепей, т. е., другими словами, нужно было бы ожидать, что свойства метаизомера будут средними между свойствами орто- и парасоединений. Это противоречие уже совершенно не объяснялось теорией и вызвало несколько новых формул строения бензола, основанием для которых было 2-ое положение Кекуле и близость свойств орто- и параизомеров. Таковыми являются формулы: Клауса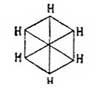 и Ланденбурга
и Ланденбурга Очевидно, что формулы В1 и В3 вытекают из B2, и потому обыкновенно принимают, что, по Ладенбургу, атомы углерода в бензольном кольце расположены не на плоскости, а в вершинах призмы, основаниями которой служат правильные треугольники и высота которой равна стороне треугольника основания. Как бы в подтверждение приведенных формул говорят исследования Томсона, который на основании термохимических данных приходит к заключению, что бензол должен иметь 9 простых связей и ни одной двойной. Но этому выводу противоречат данные Брюля относительно удельного объема (см.) бензола, которые скорее говорят за то, что он имеет 3 двойных связи; к такому же выводу приводят и работы Канонникова над лучепреломлением бензола. Однако все эти факты не являются вполне доказательными, так как еще сами отношения указанных физических свойств данного тела к его химическому строению нетвердо установлены. Ввиду этого в 90-х годах А. Байером и В. Мейером был снова предпринят целый ряд экспериментальных работ над строением аром. У., которые хотя и не привели к какой-либо определенной формуле, но находятся в согласии между собой относительно того, что аром. У. не имеют ядра с двойными связями и что четвертые (свободные) единицы сродства углеродных атомов ядра направлены симметрично к центру его;
Очевидно, что формулы В1 и В3 вытекают из B2, и потому обыкновенно принимают, что, по Ладенбургу, атомы углерода в бензольном кольце расположены не на плоскости, а в вершинах призмы, основаниями которой служат правильные треугольники и высота которой равна стороне треугольника основания. Как бы в подтверждение приведенных формул говорят исследования Томсона, который на основании термохимических данных приходит к заключению, что бензол должен иметь 9 простых связей и ни одной двойной. Но этому выводу противоречат данные Брюля относительно удельного объема (см.) бензола, которые скорее говорят за то, что он имеет 3 двойных связи; к такому же выводу приводят и работы Канонникова над лучепреломлением бензола. Однако все эти факты не являются вполне доказательными, так как еще сами отношения указанных физических свойств данного тела к его химическому строению нетвердо установлены. Ввиду этого в 90-х годах А. Байером и В. Мейером был снова предпринят целый ряд экспериментальных работ над строением аром. У., которые хотя и не привели к какой-либо определенной формуле, но находятся в согласии между собой относительно того, что аром. У. не имеют ядра с двойными связями и что четвертые (свободные) единицы сродства углеродных атомов ядра направлены симметрично к центру его; однако, как эти последние уравновешиваются между собой, указанные авторы не решают, и потому Байер предложил принимать для бензола формулу Армстронга, указывающую только на направление свободных связей и не решающую вопроса о способе их насыщения, чем эта формула и отличается от формулы Клауса. Тем не менее, из работ Байера вытекает одно важное следствие, по которому четвертые единицы сродства углеродов бензольного ядра легко изменяют свое положение, давая двойные связи. Для этого стоит только бензол или его производные перевести в гидроароматические соединения, в которых в случае неполного насыщения водородами бензольного кольца всегда наблюдается ясно выраженный характер непредельных жирных соединений. Таким образом, если бензол резко отличается по своим свойствам от жирных У., то его дигидропроизводное С6Н8, несомненно, есть жирный У. с двумя двойными связями. Этот факт в высшей степени интересен потому, что он как бы снова подтверждает теорию Кекуле о колеблющихся связях бензольного кольца, указывая на то, что эти связи едва ли занимают в нем устойчивое положение. Вся эта совокупность часто противоречащих друг другу фактов говорит о том, что до сих пор не имеется в науке достаточных средств для решения вопроса о строении бензольного ядра и, вероятно, этот вопрос вполне определенно будет разрешен в том или другом смысле только тогда, когда установится ясное представление о сущности двойной связи, так как в настоящее время химики устанавливают ее присутствие только на основании одного свойства непредельных соединений, выражающегося в их стремлении перейти в предельный тип; но весьма возможно, что это свойство в некоторых случаях парализуется другими. Подобные явления довольно часто наблюдаются в химии. Напр., известно, что гидроксилы (ОН) спиртовых и фенольных радикалов способны этерифицироваться, реагируя с йодюрами спиртов же или с ангидридами кислот; однако
однако, как эти последние уравновешиваются между собой, указанные авторы не решают, и потому Байер предложил принимать для бензола формулу Армстронга, указывающую только на направление свободных связей и не решающую вопроса о способе их насыщения, чем эта формула и отличается от формулы Клауса. Тем не менее, из работ Байера вытекает одно важное следствие, по которому четвертые единицы сродства углеродов бензольного ядра легко изменяют свое положение, давая двойные связи. Для этого стоит только бензол или его производные перевести в гидроароматические соединения, в которых в случае неполного насыщения водородами бензольного кольца всегда наблюдается ясно выраженный характер непредельных жирных соединений. Таким образом, если бензол резко отличается по своим свойствам от жирных У., то его дигидропроизводное С6Н8, несомненно, есть жирный У. с двумя двойными связями. Этот факт в высшей степени интересен потому, что он как бы снова подтверждает теорию Кекуле о колеблющихся связях бензольного кольца, указывая на то, что эти связи едва ли занимают в нем устойчивое положение. Вся эта совокупность часто противоречащих друг другу фактов говорит о том, что до сих пор не имеется в науке достаточных средств для решения вопроса о строении бензольного ядра и, вероятно, этот вопрос вполне определенно будет разрешен в том или другом смысле только тогда, когда установится ясное представление о сущности двойной связи, так как в настоящее время химики устанавливают ее присутствие только на основании одного свойства непредельных соединений, выражающегося в их стремлении перейти в предельный тип; но весьма возможно, что это свойство в некоторых случаях парализуется другими. Подобные явления довольно часто наблюдаются в химии. Напр., известно, что гидроксилы (ОН) спиртовых и фенольных радикалов способны этерифицироваться, реагируя с йодюрами спиртов же или с ангидридами кислот; однако отступают от этого общего правила, и гидроксильные группы их оксипроизводных способны реагировать с йодистым метилом СН3J только в том случае, когда они не стоят в ближайшем соседстве с группой СО. В. Мейер показал также, как сильно зависит способность кислот этерифицироваться от их строения и т. д. Одним словом, из этих примеров вытекает, что невозможно устанавливать строение какого-либо соединения, обращая внимание главным образом только на один какой-нибудь его признак, а все перечисленные работы, касающиеся строения бензола, именно и страдают односторонностью; если же взять всю совокупность данных по этому вопросу, то, по-видимому, вернее всего предположить, что или в бензоле четвертые единицы сродства действительно крайне неустойчивы по своему положению и легко, иногда даже под влиянием среды, образуют двойные связи, или же что бензол, и это, пожалуй, вернее, имеет действительно 3 двойных связи, но что непредельность их характера значительно парализована замкнутостью строения этого соединения. В таком виде представляется в настоящее время вопрос о строении бензольного ядра, а следовательно, и вообще аром. У. — Несмотря, однако, на неразрешенность этого вопроса, химия ароматических соединений очень быстро двигается вперед в других отношениях, и, конечно, свойства ароматических У., как простейших соединений этого класса, представляют для науки значительный интерес.У. аром. резко отличаются от жирных благодаря, главным образом, крайней устойчивости бензольного кольца по отношению к различным реагентам; только очень энергичными окислителями или йодистым водородом при темп. выше 280° удается разорвать это кольцо, и при этом всегда можно предположить, что предварительно здесь образуется производное гидроароматическое, которое затем уже переходит в жирное соединение. Так, по данным Марковникова и Кижнера, при восстановлении йодистым водородом бензола при высокой температуре получается метилпентаметилен
отступают от этого общего правила, и гидроксильные группы их оксипроизводных способны реагировать с йодистым метилом СН3J только в том случае, когда они не стоят в ближайшем соседстве с группой СО. В. Мейер показал также, как сильно зависит способность кислот этерифицироваться от их строения и т. д. Одним словом, из этих примеров вытекает, что невозможно устанавливать строение какого-либо соединения, обращая внимание главным образом только на один какой-нибудь его признак, а все перечисленные работы, касающиеся строения бензола, именно и страдают односторонностью; если же взять всю совокупность данных по этому вопросу, то, по-видимому, вернее всего предположить, что или в бензоле четвертые единицы сродства действительно крайне неустойчивы по своему положению и легко, иногда даже под влиянием среды, образуют двойные связи, или же что бензол, и это, пожалуй, вернее, имеет действительно 3 двойных связи, но что непредельность их характера значительно парализована замкнутостью строения этого соединения. В таком виде представляется в настоящее время вопрос о строении бензольного ядра, а следовательно, и вообще аром. У. — Несмотря, однако, на неразрешенность этого вопроса, химия ароматических соединений очень быстро двигается вперед в других отношениях, и, конечно, свойства ароматических У., как простейших соединений этого класса, представляют для науки значительный интерес.У. аром. резко отличаются от жирных благодаря, главным образом, крайней устойчивости бензольного кольца по отношению к различным реагентам; только очень энергичными окислителями или йодистым водородом при темп. выше 280° удается разорвать это кольцо, и при этом всегда можно предположить, что предварительно здесь образуется производное гидроароматическое, которое затем уже переходит в жирное соединение. Так, по данным Марковникова и Кижнера, при восстановлении йодистым водородом бензола при высокой температуре получается метилпентаметилен и вернее всего предположить, что при этой реакции сначала образуется йодистый гексил:С6Н6 + 7HJ = СН3(СН2)4—CH2J + 3J2который затем и дает уже метилпентаметилен:
и вернее всего предположить, что при этой реакции сначала образуется йодистый гексил:С6Н6 + 7HJ = СН3(СН2)4—CH2J + 3J2который затем и дает уже метилпентаметилен: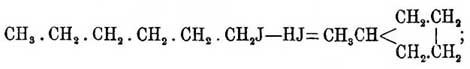 но, в свою очередь, йодистый гексил может образоваться из бензола таким образом, что в виде промежуточного продукта явится гексагидробензол C6H12, который при высокой температуре реакции присоединяет одну частицу йодистого водорода и переходит в йодистый гексил:
но, в свою очередь, йодистый гексил может образоваться из бензола таким образом, что в виде промежуточного продукта явится гексагидробензол C6H12, который при высокой температуре реакции присоединяет одну частицу йодистого водорода и переходит в йодистый гексил: При окислении бензола всегда возможно предположить образование сначала циклических производных парадигидробензола, которые затем уже переходят в открытые группировки. Далее аром. У., подобно жирным, сравнительно легко реагируют с галоидами, давая продукты замещения своих водородов галоидом; но в зависимости от условий реакции получаются при этом два рода продуктов, отличающихся друг от друга по своим свойствам. Если реакция ведется на холоду, в присутствии йода (который не способен непосредственно замещать водороды бензольного ядра), хлорной сурьмы или в крепкой серной кислоте, то получаются галоидопроизводные, в которых галоид замещает водородные атомы ядра и которые резко отличаются от соответствующих жирных соединений тем, что они не обмыливаются щелочами. В случае же, если реакция металепсии идет при кипячении У., на солнечном свете или в присутствии пятихлористого фосфора, то получается продукт, по своим свойствам не отличающийся от галоидопроизводных У. жирного ряда, и в нем галоид замещает водородные атомы боковой цепи. Из других реакций, отличающих аром. У. от жирных, необходимо указать на реакции окисления, нитрации и сульфации. Как известно, при окислении жирные У. относятся различно, в зависимости от различия их строения, У. же ароматические при более или менее энергичном окислении всегда переходят в карбоновые кислоты, при чем все их боковые углеродные цепи сгорают до образования групп CO2H, непосредственно связанных с бензольным кольцом:C6H5—CnH2n+1 + 3nO = C6H5CO2H + (n—1)CO2 + nH2O;C6H4(CnH2n+1)(CmH2m+1) + 3(n+m)O = C6H5(CO2H)2 + (n+m—2)CO2 + (m+n)H2Oи только в случае осторожного окисления марганцево-калиевою солью аром. У. с длинной боковой цепью удается проследить образование промежуточных продуктов окисления, причем из них явствует, что окисление боковой цепи совершается в этом случае совершенно по тем же правилам, как и в жирном ряду. При действии крепкой азотной кислоты на аром. У. всегда получаются нитропроизводные, в которых замещены группой NO2 водородные атомы бензольного ядра; аналогично этому и серная кислота способна при нагревании давать сульфоароматические кислоты с сульфогруппой в ядре:C6H4—CH3 + HNO3 = C6H8(NO2)—CH3 + H2OС6Н4—СН3 + Н2SO4 = С6Н3(HSO3)—СН3 + Н2О.Замечательно то, что как азотная кислота, так и серная способны заместить не более трех атомов водорода бензольного ядра и трисульфо- или тринитропроизводные совершенно не способны реагировать с указанными кислотами. В случае сульфации при этом всегда получается симметрическая трисульфокислота, в которой группы SO3H связаны или только с четными углеродными атомами, или с нечетными:
При окислении бензола всегда возможно предположить образование сначала циклических производных парадигидробензола, которые затем уже переходят в открытые группировки. Далее аром. У., подобно жирным, сравнительно легко реагируют с галоидами, давая продукты замещения своих водородов галоидом; но в зависимости от условий реакции получаются при этом два рода продуктов, отличающихся друг от друга по своим свойствам. Если реакция ведется на холоду, в присутствии йода (который не способен непосредственно замещать водороды бензольного ядра), хлорной сурьмы или в крепкой серной кислоте, то получаются галоидопроизводные, в которых галоид замещает водородные атомы ядра и которые резко отличаются от соответствующих жирных соединений тем, что они не обмыливаются щелочами. В случае же, если реакция металепсии идет при кипячении У., на солнечном свете или в присутствии пятихлористого фосфора, то получается продукт, по своим свойствам не отличающийся от галоидопроизводных У. жирного ряда, и в нем галоид замещает водородные атомы боковой цепи. Из других реакций, отличающих аром. У. от жирных, необходимо указать на реакции окисления, нитрации и сульфации. Как известно, при окислении жирные У. относятся различно, в зависимости от различия их строения, У. же ароматические при более или менее энергичном окислении всегда переходят в карбоновые кислоты, при чем все их боковые углеродные цепи сгорают до образования групп CO2H, непосредственно связанных с бензольным кольцом:C6H5—CnH2n+1 + 3nO = C6H5CO2H + (n—1)CO2 + nH2O;C6H4(CnH2n+1)(CmH2m+1) + 3(n+m)O = C6H5(CO2H)2 + (n+m—2)CO2 + (m+n)H2Oи только в случае осторожного окисления марганцево-калиевою солью аром. У. с длинной боковой цепью удается проследить образование промежуточных продуктов окисления, причем из них явствует, что окисление боковой цепи совершается в этом случае совершенно по тем же правилам, как и в жирном ряду. При действии крепкой азотной кислоты на аром. У. всегда получаются нитропроизводные, в которых замещены группой NO2 водородные атомы бензольного ядра; аналогично этому и серная кислота способна при нагревании давать сульфоароматические кислоты с сульфогруппой в ядре:C6H4—CH3 + HNO3 = C6H8(NO2)—CH3 + H2OС6Н4—СН3 + Н2SO4 = С6Н3(HSO3)—СН3 + Н2О.Замечательно то, что как азотная кислота, так и серная способны заместить не более трех атомов водорода бензольного ядра и трисульфо- или тринитропроизводные совершенно не способны реагировать с указанными кислотами. В случае сульфации при этом всегда получается симметрическая трисульфокислота, в которой группы SO3H связаны или только с четными углеродными атомами, или с нечетными: При нитрации же хотя в большинстве случаев и образуются подобные же продукты, однако, энергично нитруя 1, 4-динитробензол, удается получить и несимметрический 1, 2, 4-тринитробензол. До сих пор причина, почему не получаются при непосредственном действии азотной и серной кислот на аром. У. более чем трехзамещенные продукты, не выяснена; но существуют намеки, по крайней мере в случае азотной кислоты, что нитрогруппа (NO2) в нитропроизводных аром. У. имеет другое строение, чем приписывается ей ныне: очень может быть, что эта группа здесь трехатомна и ароматические нитротела являются производными не бензола, а дигидробензола. В самом деле, если предположить, что нитрогруппа имеет строение
При нитрации же хотя в большинстве случаев и образуются подобные же продукты, однако, энергично нитруя 1, 4-динитробензол, удается получить и несимметрический 1, 2, 4-тринитробензол. До сих пор причина, почему не получаются при непосредственном действии азотной и серной кислот на аром. У. более чем трехзамещенные продукты, не выяснена; но существуют намеки, по крайней мере в случае азотной кислоты, что нитрогруппа (NO2) в нитропроизводных аром. У. имеет другое строение, чем приписывается ей ныне: очень может быть, что эта группа здесь трехатомна и ароматические нитротела являются производными не бензола, а дигидробензола. В самом деле, если предположить, что нитрогруппа имеет строение , то тогда, принимая для бензола формулу Кекуле, возможно ход нитрации выразить следующим уравнением:
, то тогда, принимая для бензола формулу Кекуле, возможно ход нитрации выразить следующим уравнением: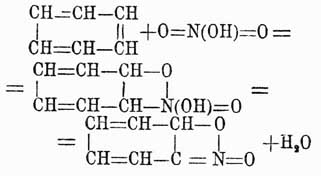 которое показывает, что более трех нитрогрупп вставить нельзя в бензольное ядро и что эти группы только и могут занимать положения 1, 3, 5 или 1, 2, 4, что и подтверждается в действительности. Хотя вышеуказанное строение NO2-группы и не подтверждено еще непосредственными наблюдениями, но в химии ароматических соединений имеется много фактов, лучше объясняемых при помощи приведенного видоизменения строения аром. нитро-У. Очевидно, что по аналогии и сульфопроизводным аром. У. можно придавать подобное же строение, и это тем более позволительно, что, вообще говоря, нитропроизводные и сульфокислоты ароматического ряда весьма резко отличаются от жирных производных, имеющих в своем составе группы NO2 и SO3H. Нитро-У. аром. при восстановлении способны давать целый ряд соединений, совершенно неизвестных в жирном ряду. Так, если восстановление это совершается в щелочной среде, то всегда наблюдаются последовательно следующие четыре реакции:
которое показывает, что более трех нитрогрупп вставить нельзя в бензольное ядро и что эти группы только и могут занимать положения 1, 3, 5 или 1, 2, 4, что и подтверждается в действительности. Хотя вышеуказанное строение NO2-группы и не подтверждено еще непосредственными наблюдениями, но в химии ароматических соединений имеется много фактов, лучше объясняемых при помощи приведенного видоизменения строения аром. нитро-У. Очевидно, что по аналогии и сульфопроизводным аром. У. можно придавать подобное же строение, и это тем более позволительно, что, вообще говоря, нитропроизводные и сульфокислоты ароматического ряда весьма резко отличаются от жирных производных, имеющих в своем составе группы NO2 и SO3H. Нитро-У. аром. при восстановлении способны давать целый ряд соединений, совершенно неизвестных в жирном ряду. Так, если восстановление это совершается в щелочной среде, то всегда наблюдаются последовательно следующие четыре реакции: Из всех этих промежуточных тел ни одного не удалось выделить в жирном ряду, где нитросоединения прямо восстановляются в амины:R—NO2 + 3H2 = RNH2 + 2H2O.Производные жирного ряда R—SO3H известны под именем сульфоновых кислот (см.) и представляют крайне непрочные соединения, легко получающиеся при действии K2SO3 на йодюры спиртов и никогда не образующиеся при непосредственном действии крепкой серной кислоты на жирные У. Сульфокислоты ароматические являются до крайности прочными телами, способными отщеплять элементы серной кислоты только при действии перегретого водяного пара при очень высокой темп. Правда, различие в свойствах сульфоновых кислот в том и другом ряду органических соединений в достаточной мере хорошо объясняется тем, что первые, т. е. жирные, суть производные сернистой кислоты, а вторые серной; но тот факт, что при действии крепкой серной кислоты на жирные У. не получается соединений, аналогичных аром. сульфокислотам, заставляет полагать, что к ним серная кислота, а также и азотная относятся несколько необычным образом. Вот, в сущности, главные характеристические черты аром. углеводородов.Что касается способов получения аромат. У., то их можно разделить на три разряда по исходным продуктам: 1) из каменноугольной смолы, 2) из соединений жирного и 3) из соединений ароматического ряда. Каменноугольная смола (см. Деготь каменноугольный) состоит, главным образом, из различных аром. У., и для выделения их из нее в чистом виде обыкновенно ее подвергают предварительно грубой дробной перегонке, отбирая 6 фракций, которые очищаются надлежащим образом и подвергаются новой, весьма тщательной дробной перегонке (см. Деготь каменноугольный). Для получения аром. У., имеющих в своем составе только одно бензольное кольцо, значение имеют исключительно первые две фракции, которые содержат бензол, толуол, ксилолы, этилбензол, триметилбензолы и дурол, или тетраметилбензол; последние У., правда, находятся и в следующей фракции, в среднем масле, но вообще их содержание в каменноугольной смоле невелико и практического значения они не имеют. Из других фракций выделяются главным образом более сложные У.: нафталин, антрацен и фенантрен. Из соединений жирного ряда аром. У. образуются при весьма разнообразных реакциях. Напр., метан СН4 при пропускании через раскаленные докрасна железные трубки конденсируется с образованием бензола. Особенно же склонны к так назыв. ароматической конденсации непредельные жирные У., некоторые их производные, кетоны, дикетоны и альдегиды. Так, ацетилен С2Н2 при краснокалильном жаре дает бензол; его производное пропиоловая кислота СН≡С—СО2Н уже на солнечном свету переходит в тримезиновую, или s-бензолтрикарбоновую, кислоту; ацетон СН3COCH3 при нагревании с серной кислотой выделяет воду и образует симметрический триметилбензол, или мезитилен; диацетил CH3CO—CO—CH3 в присутствии едкого кали конденсируется в 2,5-диметилхинон, легко дающий при восстановлении 2,5-диметилбензол, или ксилол, и т. д. Хотя в науке и известно весьма большое число реакций ароматической конденсации, т. е. таких, при которых из жирных соединений образуются ароматические, однако все они имеют только чисто теоретический интерес, так как выходы аром. У. при них очень невелики и получение их в силу этого становится и хлопотливым, и дорогим. Во всех же случаях, когда желают получить значительные количества аром. У., прибегают или к перегонке каменноугольной смолы, или же, если желаемого У. в ней не содержится, то к синтетическим реакциям, дающим возможность получить его из У. же, добываемых все-таки из каменноугольной смолы, которая, следовательно, и является, по существу, единственным исходным материалом для получения на практике аром. У. Из вышеупомянутых реакций особенного внимания заслуживают реакции Вюрца и Фриделя и Крафтса. Первая из них (Вюрца) основана на способности галоидопроизводных аром. У. и йодюров жирных спиртов в эфирном растворе конденсироваться друг с другом в присутствии металлического натрия:C6H5Br + C2H5J + 2Na = C6H5—C2H5 + NaBr + NaJ.Здесь, по-видимому, вначале происходит какое-то соединение натрия с ароматическим галоидопроизводным, обладающее темно-синим цветом, которое затем уже и реагирует с спиртовым йодюром. По крайней мере, при нормальном ходе реакции всегда натрий покрывается сначала синим налетом, превращающимся с течением времени в белую корку соли. Интересно, что такая простая на вид конденсация требует для своего выполнения соблюдения некоторых условий, которые, теоретически рассуждая, должны были бы только препятствовать ей. Напр., конденсация эта совершенно не происходит в абсолютно сухом эфирном растворе, не идет она, конечно, и в очень сыром эфире; но для нее необходим эфир, содержащий следы воды. Точно так же, как вода, облегчают реакцию следы уксусного эфира. Далее, она идет тем чище, чем более молекулярный вес жирного йодюра. Реакция Фриделя и Крафтса для своего выполнения не требует присутствия ароматического галоидного производного, и при ней спиртовый хлорюр непосредственно конденсируется с аром. У. в присутствии безводного хлористого алюминия, при чем выделяется хлористый водород:С6Н5 + СН3Cl = С6Н5—СН3 + HCl.Реакция тоже протекает, несомненно, в двух фазах: сначала образуется соединение аром. У. с хлористым алюминием, напр. AlCl3∙3C6H6, которое и реагирует дальше, и ход ее обусловливается природой жирного хлорюра. В случае, если взято монохлоропроизводное, то в зависимости от его количества по отношению к аром. У. конденсация выражается следующими уравнениями:С6Н5 + CH3Cl = C6H5CH3 + HClC6H6 + 2СН3Cl = С6Н(СН3)2 + 2HClC6H6 + 6СН3Cl = С6(СН3)6 + 6HClт. е. наблюдается образование аромат. У. с накоплением боковых жирных цепей; если берутся в реакции полихлорюры, то, наоборот, образуются аром. У. с накоплением бензольных ядер:C6H6 + CH3Cl = С6Н5СН3 + HCl2C6Н6 + CH2Cl2 = (C6H5)2CH2 + 2HCl3C6H6 + CHCl3 = (С6H5)3CH + 3HCl.Замечательно, что CCl4 в этом случае дает CH(С6H5)3, а не тетрафенилметан (С6H5)4C, который не получен до сих пор. Замещение атомов водорода бензольного кольца жирной боковой цепью при первом ходе реакции совершается более или менее правильно, именно: вторая боковая цепь большею частью становится в параположение к первой, и если это положение замещено, то в орто-. Удивительно, что хлористый алюминий способен вызывать обратную реакцию, особенно в присутствии хлористого водорода, и аром. У. с жирными боковыми цепями при кипячении с AlCl3 отщепляют жирные хлорюры, которые в свою очередь при тех же условиях могут снова конденсироваться с другими аром. У. Так, напр., пропуская сухой хлористый водород через кипящую смесь, положим, ксилола С6Н4(СН3)2 с AlCl3, наблюдается, с одной стороны, образование толуола С6Н5—СН3, бензола C6H6 и хлористого метила, а с другой три-, тетра-, пента- и гексаметилбензолов. Этот факт указывает на то, что при реакции Фриделя и Крафтса нельзя ожидать количественных выходов, так как в тот момент, когда количество синтезируемого аром. У. достигнет определенного предела, начнется обратная реакция с отщеплением от него боковых цепей; на практике обыкновенно выходы редко превышают 60—70% теоретического. Есть указания (Гольберг), что, подобно хлористому алюминию, способно возбуждать эту реакцию и безводное хлорное железо. Кроме указанных двух способов получения аром. У., часто применяются на практике еще следующие: 1) прокаливают ароматические карбоновые кислоты с негашеной или натристой известью:С6H5СО2Н + СаО = С6H6 + СаСО3.Эта реакция особенно хороша тем, что употребляемые карбоновые кисл. весьма легко иметь в совершенно чистом виде, а потому и аром. У. получаются при этом очень чистыми. 2) Обрабатывают диазосоединения при кипячении абсолютным спиртом или, еще лучше, на холоду избытком щелочного раствора закиси олова:C6H4CH3—N=N—OH + Н2 = C6H5CH3 + H2O + N2.Эта реакция имеет большое значение при решении вопросов о строении аминов, так как диазосоединения получаются при действии на амины азотистой кислоты. Следовательно, в случае неизвестности строения этого последнего цитируемая реакция определяет аром. У., от которого он произошел. Кроме того, ею пользуются и для получения гомологов бензола, сочетая ее с реакцией Гофманна, по которой хлористоводородные или сернокислые соли монозамещенных ароматических аминов с свободным орто- или параположением по отношению к NH2-группе при температуре 270—300° изомеризуются таким образом, что жирный радикал, замещающий водород аминной группы, связывается с углеродом ядра в пара- или ортоположении к амину:C6H5NH—CH3∙HCl = 1,4C6H4CH3NH2∙HCl.Обыкновенно при этом не требуется иметь монозамещенного амина как такового, а достаточно ввести в реакцию его компоненты, т. е. соль незамещенного амина и соответствующий спирт. Этим путем на фабриках получают метаксилидин, а затем и метаксилол:C6H4(CH3)NH2∙HCl + CH3OH = C6H3(CH3)2NH2∙HCl + H2Oи далее, диазотируя и восстановляя:С6Н3(СН3)2NH2∙HCl + HNO2 = C6H3(CH3)2N=N—Cl + 2H2OC6H3(CH3)2N=N—Cl + H2 = C6H4(CH3)2 + HCl + N2.3) Разлагают аром. сульфокислоты перегретым паром и серной, соляной или фосфорной кисл. при 180°:С6Н5—SO3Н + Н2О = C6H5 + H2SO4.4) Подвергают перегонке с цинковой пылью кислородсодержащие производные аром. У. Эта реакция раньше почти постоянно применялась для определения аром. У., производным которого было исследуемое вещество. Лучшие результаты она дает, когда данное кислородное производное есть кетон, альдегид или фенол. Действие цинковой пыли здесь не вполне ясно, так как трудно представить себе, откуда берется необходимый водород:C6H5CO—C6H5 + 2H2 = C6H5CH2—C6H5 + H2O.Вероятнее всего, что здесь участвует и гидрат окиси цинка, всегда имеющийся в цинковой пыли, который при высокой температуре реагирует с металлическим цинком, выделяя водород, восстановляющий уже затем ароматическое соединение:Zn(OH)2 + Zn = 2ZnO + H2.Такой взгляд подтверждается до некоторой степени тем, что смесь гидрата окиси цинка с цинковой пылью, отвечающая приведенному уравнению, значительно энергичнее восстановляет, чем свежая цинковая пыль.У. аром. представляют жидкости или твердые при обыкн. темп. тела, большею частью с характерным запахом; они нерастворимы в воде и сами являются очень хорошими растворителями для многих органических веществ. Простейший представитель аром. У. бензол C6H6 (см.) получается из каменноугольной смолы, жидок, уд. в. 0,899 при 0°, кипит при 80,36° и при 0° затвердевает в ромбических призмах, плав. при +5,42°. Бензол — один из самых важных в практическом отношении аром. У., так как его производные в большом количестве употребляются для приготовления красок, медицинских препаратов и пр. Среди продуктов животного и растительного царств находится также немало его производных. Он же послужил главным образом для развития теории ароматических соединений. Открыт бензол был в 1825 г. Фарадеем в светильном газе, полученном из масла, затем в 1834 г. Митчерлих получил его при перегонке бензойной кисл. C6H6CO2H с негашеной известью, и наконец в 1845 г. он был выделен Гофманном из каменноугольной смолы. Замещая водороды бензола жирными радикалами, можно получить различные гомологи, большинство которых находится также в газовой смоле и из которых наиболее важными являются: толуол, или метилбензол, С6Н5—СН3, жидкость уд. веса 0,871, кипящая при 110,3°. По своему характеру толуол, подобно другим гомологам, весьма похож на бензол и также весьма ценится в технике. Отличается от бензола он только тем, что имеет одну боковую цепь в виде метильной группы, поэтому он легко окисляется, давая бензойную кислоту, и имеет, кроме производных с специфическим ароматическим характером, еще производные, напоминающие соответствующие соединения жирного ряда. Так, замещая в толуоле водороды бензольного ядра, напр., хлором, гидроксилом и т. д., получают хлорюры, не способные реагировать с едкими щелочами, оксибензолы, или фенолы, отличающиеся от жирных спиртов своим более кислым характером, и пр. Наоборот, при замещении водородов метильной группы получаются и галоидопроизводные, и спирты, и кислоты и др., совершенно напоминающие тела соответствующие жирного ряда. Напр. хлористый бензил C6H5—CH2Cl легко обмыливается щелочами, давая бензильный спирт C6H5—CH2OH, a этот последний окисляется в бензойный альдегид C6H5—CHO и далее в бензойную кислоту C6H5CO2H. При обмыливании хлористого бензилидена C6H4CHCl2 сразу получается альдегид, а бензотрихлорид C6H5CCl3 в этом случае дает бензойную кислоту. Выше было указано, что дипроизводные бензола могут быть в виде 3-х изомеров, и действительно, известны три диметилбензола формулы С6Н5СН3, которые наз. орто-, пара- и метаксилолами. Все они находятся в каменноуг. смоле и различаются по следующ. свойствам: ортоксилол затвердевает при —28°, кипит при 142°, уд. вес 0,893, при окислении дает фталевую кислоту C6H4(CO2H)2 с темп. пл. 213°, при чем она распадается на воду и свой ангидрид
Из всех этих промежуточных тел ни одного не удалось выделить в жирном ряду, где нитросоединения прямо восстановляются в амины:R—NO2 + 3H2 = RNH2 + 2H2O.Производные жирного ряда R—SO3H известны под именем сульфоновых кислот (см.) и представляют крайне непрочные соединения, легко получающиеся при действии K2SO3 на йодюры спиртов и никогда не образующиеся при непосредственном действии крепкой серной кислоты на жирные У. Сульфокислоты ароматические являются до крайности прочными телами, способными отщеплять элементы серной кислоты только при действии перегретого водяного пара при очень высокой темп. Правда, различие в свойствах сульфоновых кислот в том и другом ряду органических соединений в достаточной мере хорошо объясняется тем, что первые, т. е. жирные, суть производные сернистой кислоты, а вторые серной; но тот факт, что при действии крепкой серной кислоты на жирные У. не получается соединений, аналогичных аром. сульфокислотам, заставляет полагать, что к ним серная кислота, а также и азотная относятся несколько необычным образом. Вот, в сущности, главные характеристические черты аром. углеводородов.Что касается способов получения аромат. У., то их можно разделить на три разряда по исходным продуктам: 1) из каменноугольной смолы, 2) из соединений жирного и 3) из соединений ароматического ряда. Каменноугольная смола (см. Деготь каменноугольный) состоит, главным образом, из различных аром. У., и для выделения их из нее в чистом виде обыкновенно ее подвергают предварительно грубой дробной перегонке, отбирая 6 фракций, которые очищаются надлежащим образом и подвергаются новой, весьма тщательной дробной перегонке (см. Деготь каменноугольный). Для получения аром. У., имеющих в своем составе только одно бензольное кольцо, значение имеют исключительно первые две фракции, которые содержат бензол, толуол, ксилолы, этилбензол, триметилбензолы и дурол, или тетраметилбензол; последние У., правда, находятся и в следующей фракции, в среднем масле, но вообще их содержание в каменноугольной смоле невелико и практического значения они не имеют. Из других фракций выделяются главным образом более сложные У.: нафталин, антрацен и фенантрен. Из соединений жирного ряда аром. У. образуются при весьма разнообразных реакциях. Напр., метан СН4 при пропускании через раскаленные докрасна железные трубки конденсируется с образованием бензола. Особенно же склонны к так назыв. ароматической конденсации непредельные жирные У., некоторые их производные, кетоны, дикетоны и альдегиды. Так, ацетилен С2Н2 при краснокалильном жаре дает бензол; его производное пропиоловая кислота СН≡С—СО2Н уже на солнечном свету переходит в тримезиновую, или s-бензолтрикарбоновую, кислоту; ацетон СН3COCH3 при нагревании с серной кислотой выделяет воду и образует симметрический триметилбензол, или мезитилен; диацетил CH3CO—CO—CH3 в присутствии едкого кали конденсируется в 2,5-диметилхинон, легко дающий при восстановлении 2,5-диметилбензол, или ксилол, и т. д. Хотя в науке и известно весьма большое число реакций ароматической конденсации, т. е. таких, при которых из жирных соединений образуются ароматические, однако все они имеют только чисто теоретический интерес, так как выходы аром. У. при них очень невелики и получение их в силу этого становится и хлопотливым, и дорогим. Во всех же случаях, когда желают получить значительные количества аром. У., прибегают или к перегонке каменноугольной смолы, или же, если желаемого У. в ней не содержится, то к синтетическим реакциям, дающим возможность получить его из У. же, добываемых все-таки из каменноугольной смолы, которая, следовательно, и является, по существу, единственным исходным материалом для получения на практике аром. У. Из вышеупомянутых реакций особенного внимания заслуживают реакции Вюрца и Фриделя и Крафтса. Первая из них (Вюрца) основана на способности галоидопроизводных аром. У. и йодюров жирных спиртов в эфирном растворе конденсироваться друг с другом в присутствии металлического натрия:C6H5Br + C2H5J + 2Na = C6H5—C2H5 + NaBr + NaJ.Здесь, по-видимому, вначале происходит какое-то соединение натрия с ароматическим галоидопроизводным, обладающее темно-синим цветом, которое затем уже и реагирует с спиртовым йодюром. По крайней мере, при нормальном ходе реакции всегда натрий покрывается сначала синим налетом, превращающимся с течением времени в белую корку соли. Интересно, что такая простая на вид конденсация требует для своего выполнения соблюдения некоторых условий, которые, теоретически рассуждая, должны были бы только препятствовать ей. Напр., конденсация эта совершенно не происходит в абсолютно сухом эфирном растворе, не идет она, конечно, и в очень сыром эфире; но для нее необходим эфир, содержащий следы воды. Точно так же, как вода, облегчают реакцию следы уксусного эфира. Далее, она идет тем чище, чем более молекулярный вес жирного йодюра. Реакция Фриделя и Крафтса для своего выполнения не требует присутствия ароматического галоидного производного, и при ней спиртовый хлорюр непосредственно конденсируется с аром. У. в присутствии безводного хлористого алюминия, при чем выделяется хлористый водород:С6Н5 + СН3Cl = С6Н5—СН3 + HCl.Реакция тоже протекает, несомненно, в двух фазах: сначала образуется соединение аром. У. с хлористым алюминием, напр. AlCl3∙3C6H6, которое и реагирует дальше, и ход ее обусловливается природой жирного хлорюра. В случае, если взято монохлоропроизводное, то в зависимости от его количества по отношению к аром. У. конденсация выражается следующими уравнениями:С6Н5 + CH3Cl = C6H5CH3 + HClC6H6 + 2СН3Cl = С6Н(СН3)2 + 2HClC6H6 + 6СН3Cl = С6(СН3)6 + 6HClт. е. наблюдается образование аромат. У. с накоплением боковых жирных цепей; если берутся в реакции полихлорюры, то, наоборот, образуются аром. У. с накоплением бензольных ядер:C6H6 + CH3Cl = С6Н5СН3 + HCl2C6Н6 + CH2Cl2 = (C6H5)2CH2 + 2HCl3C6H6 + CHCl3 = (С6H5)3CH + 3HCl.Замечательно, что CCl4 в этом случае дает CH(С6H5)3, а не тетрафенилметан (С6H5)4C, который не получен до сих пор. Замещение атомов водорода бензольного кольца жирной боковой цепью при первом ходе реакции совершается более или менее правильно, именно: вторая боковая цепь большею частью становится в параположение к первой, и если это положение замещено, то в орто-. Удивительно, что хлористый алюминий способен вызывать обратную реакцию, особенно в присутствии хлористого водорода, и аром. У. с жирными боковыми цепями при кипячении с AlCl3 отщепляют жирные хлорюры, которые в свою очередь при тех же условиях могут снова конденсироваться с другими аром. У. Так, напр., пропуская сухой хлористый водород через кипящую смесь, положим, ксилола С6Н4(СН3)2 с AlCl3, наблюдается, с одной стороны, образование толуола С6Н5—СН3, бензола C6H6 и хлористого метила, а с другой три-, тетра-, пента- и гексаметилбензолов. Этот факт указывает на то, что при реакции Фриделя и Крафтса нельзя ожидать количественных выходов, так как в тот момент, когда количество синтезируемого аром. У. достигнет определенного предела, начнется обратная реакция с отщеплением от него боковых цепей; на практике обыкновенно выходы редко превышают 60—70% теоретического. Есть указания (Гольберг), что, подобно хлористому алюминию, способно возбуждать эту реакцию и безводное хлорное железо. Кроме указанных двух способов получения аром. У., часто применяются на практике еще следующие: 1) прокаливают ароматические карбоновые кислоты с негашеной или натристой известью:С6H5СО2Н + СаО = С6H6 + СаСО3.Эта реакция особенно хороша тем, что употребляемые карбоновые кисл. весьма легко иметь в совершенно чистом виде, а потому и аром. У. получаются при этом очень чистыми. 2) Обрабатывают диазосоединения при кипячении абсолютным спиртом или, еще лучше, на холоду избытком щелочного раствора закиси олова:C6H4CH3—N=N—OH + Н2 = C6H5CH3 + H2O + N2.Эта реакция имеет большое значение при решении вопросов о строении аминов, так как диазосоединения получаются при действии на амины азотистой кислоты. Следовательно, в случае неизвестности строения этого последнего цитируемая реакция определяет аром. У., от которого он произошел. Кроме того, ею пользуются и для получения гомологов бензола, сочетая ее с реакцией Гофманна, по которой хлористоводородные или сернокислые соли монозамещенных ароматических аминов с свободным орто- или параположением по отношению к NH2-группе при температуре 270—300° изомеризуются таким образом, что жирный радикал, замещающий водород аминной группы, связывается с углеродом ядра в пара- или ортоположении к амину:C6H5NH—CH3∙HCl = 1,4C6H4CH3NH2∙HCl.Обыкновенно при этом не требуется иметь монозамещенного амина как такового, а достаточно ввести в реакцию его компоненты, т. е. соль незамещенного амина и соответствующий спирт. Этим путем на фабриках получают метаксилидин, а затем и метаксилол:C6H4(CH3)NH2∙HCl + CH3OH = C6H3(CH3)2NH2∙HCl + H2Oи далее, диазотируя и восстановляя:С6Н3(СН3)2NH2∙HCl + HNO2 = C6H3(CH3)2N=N—Cl + 2H2OC6H3(CH3)2N=N—Cl + H2 = C6H4(CH3)2 + HCl + N2.3) Разлагают аром. сульфокислоты перегретым паром и серной, соляной или фосфорной кисл. при 180°:С6Н5—SO3Н + Н2О = C6H5 + H2SO4.4) Подвергают перегонке с цинковой пылью кислородсодержащие производные аром. У. Эта реакция раньше почти постоянно применялась для определения аром. У., производным которого было исследуемое вещество. Лучшие результаты она дает, когда данное кислородное производное есть кетон, альдегид или фенол. Действие цинковой пыли здесь не вполне ясно, так как трудно представить себе, откуда берется необходимый водород:C6H5CO—C6H5 + 2H2 = C6H5CH2—C6H5 + H2O.Вероятнее всего, что здесь участвует и гидрат окиси цинка, всегда имеющийся в цинковой пыли, который при высокой температуре реагирует с металлическим цинком, выделяя водород, восстановляющий уже затем ароматическое соединение:Zn(OH)2 + Zn = 2ZnO + H2.Такой взгляд подтверждается до некоторой степени тем, что смесь гидрата окиси цинка с цинковой пылью, отвечающая приведенному уравнению, значительно энергичнее восстановляет, чем свежая цинковая пыль.У. аром. представляют жидкости или твердые при обыкн. темп. тела, большею частью с характерным запахом; они нерастворимы в воде и сами являются очень хорошими растворителями для многих органических веществ. Простейший представитель аром. У. бензол C6H6 (см.) получается из каменноугольной смолы, жидок, уд. в. 0,899 при 0°, кипит при 80,36° и при 0° затвердевает в ромбических призмах, плав. при +5,42°. Бензол — один из самых важных в практическом отношении аром. У., так как его производные в большом количестве употребляются для приготовления красок, медицинских препаратов и пр. Среди продуктов животного и растительного царств находится также немало его производных. Он же послужил главным образом для развития теории ароматических соединений. Открыт бензол был в 1825 г. Фарадеем в светильном газе, полученном из масла, затем в 1834 г. Митчерлих получил его при перегонке бензойной кисл. C6H6CO2H с негашеной известью, и наконец в 1845 г. он был выделен Гофманном из каменноугольной смолы. Замещая водороды бензола жирными радикалами, можно получить различные гомологи, большинство которых находится также в газовой смоле и из которых наиболее важными являются: толуол, или метилбензол, С6Н5—СН3, жидкость уд. веса 0,871, кипящая при 110,3°. По своему характеру толуол, подобно другим гомологам, весьма похож на бензол и также весьма ценится в технике. Отличается от бензола он только тем, что имеет одну боковую цепь в виде метильной группы, поэтому он легко окисляется, давая бензойную кислоту, и имеет, кроме производных с специфическим ароматическим характером, еще производные, напоминающие соответствующие соединения жирного ряда. Так, замещая в толуоле водороды бензольного ядра, напр., хлором, гидроксилом и т. д., получают хлорюры, не способные реагировать с едкими щелочами, оксибензолы, или фенолы, отличающиеся от жирных спиртов своим более кислым характером, и пр. Наоборот, при замещении водородов метильной группы получаются и галоидопроизводные, и спирты, и кислоты и др., совершенно напоминающие тела соответствующие жирного ряда. Напр. хлористый бензил C6H5—CH2Cl легко обмыливается щелочами, давая бензильный спирт C6H5—CH2OH, a этот последний окисляется в бензойный альдегид C6H5—CHO и далее в бензойную кислоту C6H5CO2H. При обмыливании хлористого бензилидена C6H4CHCl2 сразу получается альдегид, а бензотрихлорид C6H5CCl3 в этом случае дает бензойную кислоту. Выше было указано, что дипроизводные бензола могут быть в виде 3-х изомеров, и действительно, известны три диметилбензола формулы С6Н5СН3, которые наз. орто-, пара- и метаксилолами. Все они находятся в каменноуг. смоле и различаются по следующ. свойствам: ортоксилол затвердевает при —28°, кипит при 142°, уд. вес 0,893, при окислении дает фталевую кислоту C6H4(CO2H)2 с темп. пл. 213°, при чем она распадается на воду и свой ангидрид Метаксилол, или изоксилол, плавится при —54°, кипит при 139°, уд. вес 0,881, окисляется в изофталевую кислоту, неспособную давать внутренний ангидрид и плавящуюся при 300°. Параксилол плав. при 15°, кипит при 138°; уд. вес 0,880, окисляется в терефталевую кислоту, которая возгоняется, не плавясь. Изомером ксилолов является еще этилбензол С6Н5—С2Н5, кипящий при +134°, уд. вес 0,883 и получающийся синтетически по реакциям Вюрца или Фриделя и Крафтса. В отличие от ксилолов при окислении он дает бензойную кислоту, что доказывает присутствие в нем только одной боковой цепи. Следующие гомологи бензола имеют формулу С9Н12, и теоретически возможно ожидать в этом случае 8 изомеров, которые и получены в действительности. Изомеры эти следующие: 3 триметилбензола С6H3(СН3)3, 3 метилэтилбензола C6H4(CH3)(C2H5), пропилбензол С6Н5СН2СН2СН3 и изопропилбензол С6Н5CH(СН3)2. Симметрический 1, 5, 5-триметилбензол, или мезитилен, находится в каменноугольной смоле и может быть получен синтетически конденсацией ацетона (СН3)2СО или аллилена СН3C≡CH в присутствии крепкой серной кисл.3CH3СОСН3 — 3H2O = С6Н3(СН3)3.Мезитилен жидок, с довольно приятным, слегка мятным запахом, кипит при 164,5°, уд. в. 0,869. Разбавленной азотной кислотой при кипячении он постепенно окисляется таким образом, что сперва сгорает до карбоксила одна метильная группа и получается мезитиленовая кисл.
Метаксилол, или изоксилол, плавится при —54°, кипит при 139°, уд. вес 0,881, окисляется в изофталевую кислоту, неспособную давать внутренний ангидрид и плавящуюся при 300°. Параксилол плав. при 15°, кипит при 138°; уд. вес 0,880, окисляется в терефталевую кислоту, которая возгоняется, не плавясь. Изомером ксилолов является еще этилбензол С6Н5—С2Н5, кипящий при +134°, уд. вес 0,883 и получающийся синтетически по реакциям Вюрца или Фриделя и Крафтса. В отличие от ксилолов при окислении он дает бензойную кислоту, что доказывает присутствие в нем только одной боковой цепи. Следующие гомологи бензола имеют формулу С9Н12, и теоретически возможно ожидать в этом случае 8 изомеров, которые и получены в действительности. Изомеры эти следующие: 3 триметилбензола С6H3(СН3)3, 3 метилэтилбензола C6H4(CH3)(C2H5), пропилбензол С6Н5СН2СН2СН3 и изопропилбензол С6Н5CH(СН3)2. Симметрический 1, 5, 5-триметилбензол, или мезитилен, находится в каменноугольной смоле и может быть получен синтетически конденсацией ацетона (СН3)2СО или аллилена СН3C≡CH в присутствии крепкой серной кисл.3CH3СОСН3 — 3H2O = С6Н3(СН3)3.Мезитилен жидок, с довольно приятным, слегка мятным запахом, кипит при 164,5°, уд. в. 0,869. Разбавленной азотной кислотой при кипячении он постепенно окисляется таким образом, что сперва сгорает до карбоксила одна метильная группа и получается мезитиленовая кисл. затем окисление идет дальше, и образуется увитиновая кисл.
затем окисление идет дальше, и образуется увитиновая кисл. и, наконец, тримезиновая кислота 1, 3, 5-C6H3(CO2H)3.Несимметрический 1,3,4-триметилбензол, или псевдокумол, т. кип. 170°, также находится в каменноугольной смоле и отделяется от мезитилена переводом смеси этих У. в моносульфокислоты, для чего 1 объем сырого псевдокумола дигерируют при 80—90° с одним объемом обыкновенной серной кислоты. При этом часть вещества растворяется в кислоте, часть же всплывает наверх в виде маслянистого слоя, который отбрасывают, а к кислому раствору прибавляют понемногу 1/3 об. воды и оставляют стоять на 24 часа, после чего жидкость снова разделяется на два слоя — нижний, состоящий из разбавленной серной кислоты, и верхний — из сульфокислот. Нижний слой удаляется, а к верхнему прибавляют еще около 1/3 об. воды и нагревают до получения совершенно прозрачного раствора, из которого при охлаждении кристаллизуется труднорастворимая псевдокумолсульфоновая кисл., соответствующая же кисл. мезитилена остается в растворе.Рядовой 1, 2, 3-триметилбензол, или гемимеллитол, темп. кип. 175°, находится вместе с предыдущими двумя изомерами. Из других изомеров формулы С9Н12 заслуживает упоминания только кумол, или изопропилбензол, С6H5СН(СН3)2 (темп. кип. 153°), который в первый раз был получен при перегон. с известью куминовой кислоты пара-(СН3)2СН—С6Н4—СО2Н, получающейся при окислении тминного масла. Синтетически этот У. может быть получен из C6H5CHCl2 и цинкметила Zn(СН3)2, а также при нагревании бензина с бромистым нормальн. пропилом или изопропилом в присутствии хлористого алюминия; интересно, что под влиянием, вероятно, AlCl3 или HCl при этой реакции норм. бромистый пропил изомеризуется в изосоединение, так что этим методом невозможно получить норм. пропилбензол, который легко образуется по реакции Вюрца. Кумол важен в том отношении, что он послужил для установки строения пропильной группы его гомолога цимола (см. ниже), которая раньше считалась нормальной, и только в 1891 г. Видман показал, что кумол, а следовательно, и цимол, легко окисляющийся в куминовую кислоту, имеет в своем составе изопропильную группу. Цимол, или параметилкумол (СН3)2СН—СбН4—СН3, темп. кип. 153°, имеет большое значение, так как его производные в очень многих случаях являются главными составными частями растительных масел, бальзамов, смол и пр. Точно так же важнейшие терпены имеют в своем составе гидрогенизированное цимоловое кольцо. Этот У. и получается при нагревании камфоры с фосфорным ангидридом.Из тетраметилбензолов С6H2(CH3)4 интересен только симметрический 1, 2, 4, 5-, или дурол, так как это первый гомолог бензола, твердый при обыкн. темп. Он плавится при 79° и кипит при 190°. Пентаметилбензол плавится при 53° и кипит при 230°. Гексаметилбензол плавится при 164° и кипит при 264°. Последние два У. получаются синтетически, по реакции Фриделя и Крафтса, из толуола или других метилбензолов при конденсации их с хлористым метилом CH3Cl. Гексаметилбензол при окислении дает бензолгексакарбоновую, или меллитовую, кислоту С6(СО2Н)6 (см.) и интересен тем, что в нем все водороды бензольного кольца замещены боковыми жирными цепями, а потому он может давать производные только одного типа, по своим свойствам аналогичные жирным, и, следовательно, его ароматический характер сильно маскирован; так, напр., с азотной или серной кислотами он реагирует как жирный У. и не дает нитро- или сульфопроизводных.При замещении водородов бензола непредельными жирными радикалами получаются ароматич. углеводороды, обладающие двойственным характером ароматических и непредельных жирных соединений. Напр., при конденсации бромистого винила с бензолом получается фенилэтилен, или стирол, C6H5—CH=CH2, темп. кип. 144°. Этот У. находится в количестве 1—5% в свободном состоянии в стираксе (см. Смолы) и легче всего может быть получен из коричной кислоты С6Н5—СН=СНСО2Н, которая, подобно жирным непредельным соединениям, легко присоединяет бромистый водород и переходит в β-бромгидрокоричную кислоту СбН5CHBr—CH2—СО2Н, а эта последняя, в свою очередь, при нагревании с раствором соды количественно распадается на HBr, CO2 и стирол:С6H5CHBrCH2—СО2Н = С6Н5СН=СН2 + HBr + CO2.Стирол весьма легко присоединяет галоиды, образуя дигалоидные производные этилбензола С6Н5СНХ—СН2Х; с галоидоводородными кислотами он дает соединения типа С6H5СНХ—СН3 и С6H5СН2—СН2Х и т. д., и в то же время известны его нитро- и амидопроизводные совершенно ароматического характера с замещенными водородами в ядре NO2C6H4—CH=CH2 и др. С другой стороны, C6H5CBr=CH2, подобно бромюрам жирных У., от действия спиртового раствора едкого кали отщепляет HBr и дает фенилацетилен C6H5C≡CH, обладающий всеми характерными свойствами У. ацетиленовых.До сих пор были рассмотрены У., содержащие одно бензольное кольцо; но, подобно тому, как в предыдущих примерах водороды кольца замещались жирными радикалами, их можно замещать и ароматическими, точно так же возможно и водороды жирных У. замещать остатками У. аром. В первом случае получаются соединения типа C6H5—C6H5 (дифенилбензол [Группа С6Н5 обыкновенно называется фенилом.]), С6Н5—С6H4СН3 (фенилтолил), СбН4(С6Н5)2 (дифенилбензол) и пр., во втором случае образуются CH2(C6H5)2 (дифенилметан), (C6H5)3CH (трифенилметан), C6H5—CH2—CH2C6H5 (дибензил) и пр. Однако в этих случаях конденсации идут значительно труднее, и, например, до сих пор еще не получено полифенилбензолов, в которых бы было замещено более трех водородов бензольного ядра фенилами; неизвестен и тетрафенилметан, пентафенилэтан и пр. Вообще, кажется, замещение водородов жирных предельных У. фенильными группами идет с накоплением в частиц этих последних до тех пор, пока при каждом углероде жирного соединения не останется по одному водороду, и после этого момента конденсация уже не совершается.Простейший У. первого типа, дифенил C6H5—C6H5 может быть получен или по реакции Вюрца из С6Н5Br и Na, или же, что проще, при пропускании паров бензола через докрасна раскаленные железные трубки. Дифенил уже тверд при обыкновенной температуре, он кристаллизуется из спирта в больших бесцветных таблицах с темп. пл. 71° и темп. кип. 254°. При окислении хромовой кислотой он, подобно всем аром. У. с одной боковой цепью, дает бензойную кислоту C6H5—CO2H, и это окисление идет легче окисления бензола, несмотря на то, что дифенил содержит только бензольные ядра. Здесь как будто бы реакция облегчается в силу образования конечного продукта, принадлежащего к ароматическому же ряду, а не к жирному, как это имеет место при окислении бензола, потому что такое уменьшение прочности бензольного ядра по отношению к окислителям наблюдается всякий раз, как только окончательным продуктом является ароматическое же соединение. Напр. нафталин (см. ниже) сравнительно легко окисляется в фталевую кисл., фенантрен (см. ниже) в дифеновую и т. д. Многие производные дифенила образуют отличные краски; но эти производные редко получаются из самого У., обыкновенно же пользуются для этой цели способностью гидразосоединений RNH—NHR' (где R и R' остатки каких-либо аром. У.) в присутствии минеральных кислот изомеризоваться в амидопроизводные дибензола и его гомологов:C6H5NH—NHC6H4CH3 = NH2C6H4—C6H3(CH3)NH2причем всегда, если свободно параместо по отношению к амидогруппе, то дифенильная связь и занимает его; в случае, если параместо занято, то связь смыкается в ортоположении, если же и это место несвободно, то реакция изомеризации не идет. Эта же реакция дает возможность получить и различные гомологи дифенила, исходя из гомологов нитробензола, переводя последние в гидразосоединения. Так, гидразобензол
и, наконец, тримезиновая кислота 1, 3, 5-C6H3(CO2H)3.Несимметрический 1,3,4-триметилбензол, или псевдокумол, т. кип. 170°, также находится в каменноугольной смоле и отделяется от мезитилена переводом смеси этих У. в моносульфокислоты, для чего 1 объем сырого псевдокумола дигерируют при 80—90° с одним объемом обыкновенной серной кислоты. При этом часть вещества растворяется в кислоте, часть же всплывает наверх в виде маслянистого слоя, который отбрасывают, а к кислому раствору прибавляют понемногу 1/3 об. воды и оставляют стоять на 24 часа, после чего жидкость снова разделяется на два слоя — нижний, состоящий из разбавленной серной кислоты, и верхний — из сульфокислот. Нижний слой удаляется, а к верхнему прибавляют еще около 1/3 об. воды и нагревают до получения совершенно прозрачного раствора, из которого при охлаждении кристаллизуется труднорастворимая псевдокумолсульфоновая кисл., соответствующая же кисл. мезитилена остается в растворе.Рядовой 1, 2, 3-триметилбензол, или гемимеллитол, темп. кип. 175°, находится вместе с предыдущими двумя изомерами. Из других изомеров формулы С9Н12 заслуживает упоминания только кумол, или изопропилбензол, С6H5СН(СН3)2 (темп. кип. 153°), который в первый раз был получен при перегон. с известью куминовой кислоты пара-(СН3)2СН—С6Н4—СО2Н, получающейся при окислении тминного масла. Синтетически этот У. может быть получен из C6H5CHCl2 и цинкметила Zn(СН3)2, а также при нагревании бензина с бромистым нормальн. пропилом или изопропилом в присутствии хлористого алюминия; интересно, что под влиянием, вероятно, AlCl3 или HCl при этой реакции норм. бромистый пропил изомеризуется в изосоединение, так что этим методом невозможно получить норм. пропилбензол, который легко образуется по реакции Вюрца. Кумол важен в том отношении, что он послужил для установки строения пропильной группы его гомолога цимола (см. ниже), которая раньше считалась нормальной, и только в 1891 г. Видман показал, что кумол, а следовательно, и цимол, легко окисляющийся в куминовую кислоту, имеет в своем составе изопропильную группу. Цимол, или параметилкумол (СН3)2СН—СбН4—СН3, темп. кип. 153°, имеет большое значение, так как его производные в очень многих случаях являются главными составными частями растительных масел, бальзамов, смол и пр. Точно так же важнейшие терпены имеют в своем составе гидрогенизированное цимоловое кольцо. Этот У. и получается при нагревании камфоры с фосфорным ангидридом.Из тетраметилбензолов С6H2(CH3)4 интересен только симметрический 1, 2, 4, 5-, или дурол, так как это первый гомолог бензола, твердый при обыкн. темп. Он плавится при 79° и кипит при 190°. Пентаметилбензол плавится при 53° и кипит при 230°. Гексаметилбензол плавится при 164° и кипит при 264°. Последние два У. получаются синтетически, по реакции Фриделя и Крафтса, из толуола или других метилбензолов при конденсации их с хлористым метилом CH3Cl. Гексаметилбензол при окислении дает бензолгексакарбоновую, или меллитовую, кислоту С6(СО2Н)6 (см.) и интересен тем, что в нем все водороды бензольного кольца замещены боковыми жирными цепями, а потому он может давать производные только одного типа, по своим свойствам аналогичные жирным, и, следовательно, его ароматический характер сильно маскирован; так, напр., с азотной или серной кислотами он реагирует как жирный У. и не дает нитро- или сульфопроизводных.При замещении водородов бензола непредельными жирными радикалами получаются ароматич. углеводороды, обладающие двойственным характером ароматических и непредельных жирных соединений. Напр., при конденсации бромистого винила с бензолом получается фенилэтилен, или стирол, C6H5—CH=CH2, темп. кип. 144°. Этот У. находится в количестве 1—5% в свободном состоянии в стираксе (см. Смолы) и легче всего может быть получен из коричной кислоты С6Н5—СН=СНСО2Н, которая, подобно жирным непредельным соединениям, легко присоединяет бромистый водород и переходит в β-бромгидрокоричную кислоту СбН5CHBr—CH2—СО2Н, а эта последняя, в свою очередь, при нагревании с раствором соды количественно распадается на HBr, CO2 и стирол:С6H5CHBrCH2—СО2Н = С6Н5СН=СН2 + HBr + CO2.Стирол весьма легко присоединяет галоиды, образуя дигалоидные производные этилбензола С6Н5СНХ—СН2Х; с галоидоводородными кислотами он дает соединения типа С6H5СНХ—СН3 и С6H5СН2—СН2Х и т. д., и в то же время известны его нитро- и амидопроизводные совершенно ароматического характера с замещенными водородами в ядре NO2C6H4—CH=CH2 и др. С другой стороны, C6H5CBr=CH2, подобно бромюрам жирных У., от действия спиртового раствора едкого кали отщепляет HBr и дает фенилацетилен C6H5C≡CH, обладающий всеми характерными свойствами У. ацетиленовых.До сих пор были рассмотрены У., содержащие одно бензольное кольцо; но, подобно тому, как в предыдущих примерах водороды кольца замещались жирными радикалами, их можно замещать и ароматическими, точно так же возможно и водороды жирных У. замещать остатками У. аром. В первом случае получаются соединения типа C6H5—C6H5 (дифенилбензол [Группа С6Н5 обыкновенно называется фенилом.]), С6Н5—С6H4СН3 (фенилтолил), СбН4(С6Н5)2 (дифенилбензол) и пр., во втором случае образуются CH2(C6H5)2 (дифенилметан), (C6H5)3CH (трифенилметан), C6H5—CH2—CH2C6H5 (дибензил) и пр. Однако в этих случаях конденсации идут значительно труднее, и, например, до сих пор еще не получено полифенилбензолов, в которых бы было замещено более трех водородов бензольного ядра фенилами; неизвестен и тетрафенилметан, пентафенилэтан и пр. Вообще, кажется, замещение водородов жирных предельных У. фенильными группами идет с накоплением в частиц этих последних до тех пор, пока при каждом углероде жирного соединения не останется по одному водороду, и после этого момента конденсация уже не совершается.Простейший У. первого типа, дифенил C6H5—C6H5 может быть получен или по реакции Вюрца из С6Н5Br и Na, или же, что проще, при пропускании паров бензола через докрасна раскаленные железные трубки. Дифенил уже тверд при обыкновенной температуре, он кристаллизуется из спирта в больших бесцветных таблицах с темп. пл. 71° и темп. кип. 254°. При окислении хромовой кислотой он, подобно всем аром. У. с одной боковой цепью, дает бензойную кислоту C6H5—CO2H, и это окисление идет легче окисления бензола, несмотря на то, что дифенил содержит только бензольные ядра. Здесь как будто бы реакция облегчается в силу образования конечного продукта, принадлежащего к ароматическому же ряду, а не к жирному, как это имеет место при окислении бензола, потому что такое уменьшение прочности бензольного ядра по отношению к окислителям наблюдается всякий раз, как только окончательным продуктом является ароматическое же соединение. Напр. нафталин (см. ниже) сравнительно легко окисляется в фталевую кисл., фенантрен (см. ниже) в дифеновую и т. д. Многие производные дифенила образуют отличные краски; но эти производные редко получаются из самого У., обыкновенно же пользуются для этой цели способностью гидразосоединений RNH—NHR' (где R и R' остатки каких-либо аром. У.) в присутствии минеральных кислот изомеризоваться в амидопроизводные дибензола и его гомологов:C6H5NH—NHC6H4CH3 = NH2C6H4—C6H3(CH3)NH2причем всегда, если свободно параместо по отношению к амидогруппе, то дифенильная связь и занимает его; в случае, если параместо занято, то связь смыкается в ортоположении, если же и это место несвободно, то реакция изомеризации не идет. Эта же реакция дает возможность получить и различные гомологи дифенила, исходя из гомологов нитробензола, переводя последние в гидразосоединения. Так, гидразобензол и т. д. Все полученные таким образом диамидопроизводные дифенила легко диазотируются и затем при стоянии с щелочным раствором закиси олова разлагаются с выделением азота и образованием дифенила и его гомологов. В ближайшей же генетической связи с дифенилом стоит и карбазол (см.). Дифенилбензолов C6H4(C6H5)2 известно два, мета- или изодифенилбензол, темп. пл. 85°, темп. кип. 369°, и парадифенилбензол, темп. пл. 205°, темп. кип. 383°. Получаются они или по реакции Вюрца из бромбензола и пара- или метадибромбензола, или же вместе с дифенилом при пропускании паров бензола через раскаленную трубку. Трифенилбензолов С6Н5(С6Н5)3 получено тоже только два; из них один с не вполне выясненным строением, а другой симметрический 1, 3, 5, темп. пл. 169°, получающийся из ацетофенона С6Н5СОСН3, подобно тому как мезитилен (см. выше) получается из ацетона:3СбН5СОСН3 = С6Н3(С6H5)3 + 3Н2О.Ко второму типу ароматических углеводородов с несколькими бензольными ядрами принадлежит дифенилметан CH2(C6H5)2, получающийся проще всего по реакции Фриделя и Крафтса из бензола и хлористого бензила С6Н5CH2Cl. У. кристаллизуется в длинных призмах, плав. при 26° и кип. при 261°; он имеет запах апельсинных корок. Интересен только в теоретическом отношении, так как имеет в числе своих производных некоторые краски. При окислении дает кетон С6Н5СО—С6Н5, известный под назв. бензофенона, который, подобно жирным кетонам, при восстановлении переходит во вторичный спирт дифенилкарбинол, или бензгидрол (С6Н5)2СНОН, а при сплавлении с едким кали распадается на бензойную кислоту и бензол:C6H5COC6H5 + H2O = C6H5—COOH + C6H6.Следующий гомолог дифенилметана, трифенилметан СН(С6Н5)3, был открыт Кекуле и Франшимоном в 1872 г. в продуктах реакции хлористого бензилидена C6H5CHCl2 на меркурдифенил Hg(C6H5)2, легче же всего он получается по Фриделю и Крафтсу из хлороформа CHCl3 и бензола. Реакция эта идет очень гладко, и получающийся сырой трифенилметан легко очищается кристаллизацией из горячего бензола, с которым он образует молекулярное соединение СН(С6Н5)3∙С6Н6, чрезвычайно трудно растворимое в холодном бензоле. Для получения чистого трифенилметана указанное его соединение с бензолом нагревают на водяной бане до удаления всего бензола и полученную сплавленную массу У. кристаллизуют из спирта. Трифенилметан плавится при 92° и кипит при 358°. В этом У. в силу накопления большого количества фенильных групп, имеющих несколько отрицательный характер, водород жирного радикала приобрел свойство замещаться щелочными металлами, и при сплавлении, напр., трифенилметана с металлическим калием образуется соль (C6H5)CK, которая легко разлагается водой на свои компоненты, а при действии на нее угольного ангидрида переходит в соль трифенилуксусной кислоты (С6Н5)3C—CO2Н. Трифенилметан играл очень крупную роль при установке строения фуксина и его аналогов, и до настоящего времени трифенилметановые пигменты (см. Розанилин, Фенолфталеин, Флуоресцеин, Фуксин, Эозин, Розоловая кисл. и пр.) еще выделяются в особый класс красок, строение которых не вполне определено. Все эти краски происходят от третичного спирта трифенилкарбинола (C6H5)3C—OH, легко получающегося окислением трифенилметана.Подобно тому, как в метане можно замещать водороды фенильными группами, их можно замещать и в других жирных углеводородах, при этом, теоретически рассуждая, должны образоваться весьма разнообразные ароматические У., но на практике получены только весьма немногие из них. Среди производных этана известны асимметрические — типа R2CH—CH3, и симметрические, в которых фенильные группы замещают одинаковое число атомов водорода при каждом углероде. Для других же жирных У. известны только дифенильные производные ω1ω-ряда, т. е. производные нормальных У., в которых замещено фенилом по одному водороду у крайних углеродов, напр., дифенилпропан C6H5—CH2—CH2—CH2—C6H5, дифенилбутан СбН5(СН2)4—C6H5 и т. д.as-Дифенилэтан (C6H5)2CH—CH3, темп. кип. 269°, получается конденсацией на холоду бензола с паральдегидом в присутствии серной кислоты или по реакции Фриделя и Крафтса из 5-бромэтилбензола C6H5CHBrCH3 и бензола. При окислении хромовой кислотой этот У. отщепляет метильную группу и переходит в бензофенон, если же окислять его азотной кислотой, то сначала получаются продукты нитрации, причем удивительно, что в этом случае бензольные ядра не нитруются, а подвергается действию азотной кислоты опять-таки жирный радикал. Однако в этом случае получаются не истинные нитротела, а азотистые эфиры дифенилэтиленгликоля (C6H5)2C(OH)—CH2ONO (темп. пл. 100°) дифенилвинильного спирта (C6H5)2C=CHONO (темп. пл. 86°) и эфир с темп. пл. 148—149°, содержащей две ONO-группы, но строение которого ближе не исследовано. Такое отступление от общего правила действия HNO3 на аром. У. можно только объяснить тем, что в данном случае жирный радикал окисляется сначала в дифенлэтиленгликоль (C6H5)2C(ОН), а этот последний, подобно всем жирным многоатомным спиртам, весьма легко с азотной кислотой образует азотистый эфир. Образование же эфира дифенилвинильного спирта легко объяснить отщеплением воды от предыдущего соединения:(C6H5)2—C(ОН)—CH2ONO — H2О = (С6Н5)C=CH—ONO.При конденсации на холоду в присутствии серной кислоты бензола с моно-, ди- и трихлоральдегидом легко получаются соответствующие хлоропроизводные дифенилэтана: (C6H5)2CH—CH2Cl—(С6Н5)2CH—CHCl2 и (C6H5)2CH—CCl3; из них монохлордифенилэтан, нагретый до начала разложения, теряет HCl и переходит не в as-дифенилэтилен, как это можно было бы ожидать по уравн.:(C6H5)2CHCH2Cl — HCl = (C6H5)2C=CH2а в его симметрический изомер, называемый стильбеном (C6H5)2CH=CH(C6H5)2 (см. ниже); той же самой изомеризации с образованием стильбена подвергаются и два другие хлорюра при восстановлении их цинковой пылью. Точно так же as-дифенилэтилен (C6H5)2C=CH2, легко получающийся обмыливанием едким кали вышеуказанного монохлордифенилэтана, дает и монохлоропроизводное (C6H5)2C=CHCl, которое при нагревании с спиртовым раствором этилата натрия рядом с эфиром дифенилвинильного спирта дает толан C6H5—C≡C—C6H5 (см. ниже). Образование этого последнего аром. У. до некоторой степени объясняет ход такой странной изомеризации: очевидно, в этих случаях водород для образования хлористого водорода берется не из жирного радикала, а из бензольного ядра и в первую фазу реакции получается продукт
и т. д. Все полученные таким образом диамидопроизводные дифенила легко диазотируются и затем при стоянии с щелочным раствором закиси олова разлагаются с выделением азота и образованием дифенила и его гомологов. В ближайшей же генетической связи с дифенилом стоит и карбазол (см.). Дифенилбензолов C6H4(C6H5)2 известно два, мета- или изодифенилбензол, темп. пл. 85°, темп. кип. 369°, и парадифенилбензол, темп. пл. 205°, темп. кип. 383°. Получаются они или по реакции Вюрца из бромбензола и пара- или метадибромбензола, или же вместе с дифенилом при пропускании паров бензола через раскаленную трубку. Трифенилбензолов С6Н5(С6Н5)3 получено тоже только два; из них один с не вполне выясненным строением, а другой симметрический 1, 3, 5, темп. пл. 169°, получающийся из ацетофенона С6Н5СОСН3, подобно тому как мезитилен (см. выше) получается из ацетона:3СбН5СОСН3 = С6Н3(С6H5)3 + 3Н2О.Ко второму типу ароматических углеводородов с несколькими бензольными ядрами принадлежит дифенилметан CH2(C6H5)2, получающийся проще всего по реакции Фриделя и Крафтса из бензола и хлористого бензила С6Н5CH2Cl. У. кристаллизуется в длинных призмах, плав. при 26° и кип. при 261°; он имеет запах апельсинных корок. Интересен только в теоретическом отношении, так как имеет в числе своих производных некоторые краски. При окислении дает кетон С6Н5СО—С6Н5, известный под назв. бензофенона, который, подобно жирным кетонам, при восстановлении переходит во вторичный спирт дифенилкарбинол, или бензгидрол (С6Н5)2СНОН, а при сплавлении с едким кали распадается на бензойную кислоту и бензол:C6H5COC6H5 + H2O = C6H5—COOH + C6H6.Следующий гомолог дифенилметана, трифенилметан СН(С6Н5)3, был открыт Кекуле и Франшимоном в 1872 г. в продуктах реакции хлористого бензилидена C6H5CHCl2 на меркурдифенил Hg(C6H5)2, легче же всего он получается по Фриделю и Крафтсу из хлороформа CHCl3 и бензола. Реакция эта идет очень гладко, и получающийся сырой трифенилметан легко очищается кристаллизацией из горячего бензола, с которым он образует молекулярное соединение СН(С6Н5)3∙С6Н6, чрезвычайно трудно растворимое в холодном бензоле. Для получения чистого трифенилметана указанное его соединение с бензолом нагревают на водяной бане до удаления всего бензола и полученную сплавленную массу У. кристаллизуют из спирта. Трифенилметан плавится при 92° и кипит при 358°. В этом У. в силу накопления большого количества фенильных групп, имеющих несколько отрицательный характер, водород жирного радикала приобрел свойство замещаться щелочными металлами, и при сплавлении, напр., трифенилметана с металлическим калием образуется соль (C6H5)CK, которая легко разлагается водой на свои компоненты, а при действии на нее угольного ангидрида переходит в соль трифенилуксусной кислоты (С6Н5)3C—CO2Н. Трифенилметан играл очень крупную роль при установке строения фуксина и его аналогов, и до настоящего времени трифенилметановые пигменты (см. Розанилин, Фенолфталеин, Флуоресцеин, Фуксин, Эозин, Розоловая кисл. и пр.) еще выделяются в особый класс красок, строение которых не вполне определено. Все эти краски происходят от третичного спирта трифенилкарбинола (C6H5)3C—OH, легко получающегося окислением трифенилметана.Подобно тому, как в метане можно замещать водороды фенильными группами, их можно замещать и в других жирных углеводородах, при этом, теоретически рассуждая, должны образоваться весьма разнообразные ароматические У., но на практике получены только весьма немногие из них. Среди производных этана известны асимметрические — типа R2CH—CH3, и симметрические, в которых фенильные группы замещают одинаковое число атомов водорода при каждом углероде. Для других же жирных У. известны только дифенильные производные ω1ω-ряда, т. е. производные нормальных У., в которых замещено фенилом по одному водороду у крайних углеродов, напр., дифенилпропан C6H5—CH2—CH2—CH2—C6H5, дифенилбутан СбН5(СН2)4—C6H5 и т. д.as-Дифенилэтан (C6H5)2CH—CH3, темп. кип. 269°, получается конденсацией на холоду бензола с паральдегидом в присутствии серной кислоты или по реакции Фриделя и Крафтса из 5-бромэтилбензола C6H5CHBrCH3 и бензола. При окислении хромовой кислотой этот У. отщепляет метильную группу и переходит в бензофенон, если же окислять его азотной кислотой, то сначала получаются продукты нитрации, причем удивительно, что в этом случае бензольные ядра не нитруются, а подвергается действию азотной кислоты опять-таки жирный радикал. Однако в этом случае получаются не истинные нитротела, а азотистые эфиры дифенилэтиленгликоля (C6H5)2C(OH)—CH2ONO (темп. пл. 100°) дифенилвинильного спирта (C6H5)2C=CHONO (темп. пл. 86°) и эфир с темп. пл. 148—149°, содержащей две ONO-группы, но строение которого ближе не исследовано. Такое отступление от общего правила действия HNO3 на аром. У. можно только объяснить тем, что в данном случае жирный радикал окисляется сначала в дифенлэтиленгликоль (C6H5)2C(ОН), а этот последний, подобно всем жирным многоатомным спиртам, весьма легко с азотной кислотой образует азотистый эфир. Образование же эфира дифенилвинильного спирта легко объяснить отщеплением воды от предыдущего соединения:(C6H5)2—C(ОН)—CH2ONO — H2О = (С6Н5)C=CH—ONO.При конденсации на холоду в присутствии серной кислоты бензола с моно-, ди- и трихлоральдегидом легко получаются соответствующие хлоропроизводные дифенилэтана: (C6H5)2CH—CH2Cl—(С6Н5)2CH—CHCl2 и (C6H5)2CH—CCl3; из них монохлордифенилэтан, нагретый до начала разложения, теряет HCl и переходит не в as-дифенилэтилен, как это можно было бы ожидать по уравн.:(C6H5)2CHCH2Cl — HCl = (C6H5)2C=CH2а в его симметрический изомер, называемый стильбеном (C6H5)2CH=CH(C6H5)2 (см. ниже); той же самой изомеризации с образованием стильбена подвергаются и два другие хлорюра при восстановлении их цинковой пылью. Точно так же as-дифенилэтилен (C6H5)2C=CH2, легко получающийся обмыливанием едким кали вышеуказанного монохлордифенилэтана, дает и монохлоропроизводное (C6H5)2C=CHCl, которое при нагревании с спиртовым раствором этилата натрия рядом с эфиром дифенилвинильного спирта дает толан C6H5—C≡C—C6H5 (см. ниже). Образование этого последнего аром. У. до некоторой степени объясняет ход такой странной изомеризации: очевидно, в этих случаях водород для образования хлористого водорода берется не из жирного радикала, а из бензольного ядра и в первую фазу реакции получается продукт затем снова происходит присоединение HCl по уравнению:
затем снова происходит присоединение HCl по уравнению: и наконец этот последний хлорюр дает нормально толан:C6H5—CCl=CH—C6H5 — HCl = C6H5С=CC6H5[Очевидно, совершенно такое же толкование реакции можно применить и для случая образования стильбена из монохлордифенилэтана.].В ходе этих реакций в сторону образования симметрических соединений наблюдается некоторая аналогия с образованием симметрических ацетиленовых углеводородов жирного ряда из их несимметрических изомеров под влиянием едкого натра при высокой температуре. Реакцию эту изучал Фаворский и показал, что здесь всегда идет изомеризация в сторону образования наиболее симметрического продукта; то же самое мы наблюдаем и в вышеприведенных случаях, причем конечным результатом и здесь являются симметричный дифенилацетилен или дифенилэтилен. Ввиду такой аналогии возможно предположить вообще в производных этилена и ацетилена стремление образовать наиболее симметричные продукты, и в этом случае было бы интересно попробовать изомеризовать as-дифенилэтилен и дифенилацетилен в условиях реакции Фаворского. s-Дифенилэтан, или дибензил С6Н5—СН2—СН2—С6Н5, темп. пл. 52°, темп. кип. 284, получается обыкновенными синтетическими реакциями из C6Н5CH2Cl или восстановлением йодистоводородной кисл. бензоина C6H5CH(OH)—CO—C6H5, который, в свою очередь, легко образуется конденсацией бензойного альдегида в присутствии цианистого калия:C6H5CHO + OCH—C6H5 = C6H5CH(OH)—COC6H5.При нагревании до 500° дибензил претерпевает характерное разложение с выделением водорода, и в этом случае образуются стильбен, толан и фенантрен (см. ниже); при окислении же дибензил распадается, давая бензойную кислоту.s-Дифенилэтилен, или стильбен C6H5CH=CHC6H5, темп. пл. 124°, темп. кип. 306°, получается, кроме вышеуказанной реакции разложения дибензила и обыкновенных синтетических реакций, еще при многих других превращениях различных ароматических соединений, и потому он принадлежит к весьма давно открытым аром. У. В первый раз его получил Лоран в 1844 г. Между прочим, интересно его образование из дифенильного эфира фумаровой кислоты C6H5OCOCH=CH—COOC6H5, который при перегонке распадается на углекислоту и стильбен:C6H5OCO—CH=CH—COOC6H5 = 2CO2 + C6H5CH=CH—C6H5.Этот У., подобно этилену, способен присоединять 2 атома брома и давать s-дифенилдибромэтан C6H5Br—CHBr—C6H5, который, в свою очередь, при обмыливании спиртовым раствором едкого кали дает дифенилацетилен, или толан, C6H5C≡CC6H5, темп. пл. 60°.Трифенилэтан (C6H5)2CH—CH2C6H5 до сих пор не получен; но известны некоторые его производные.Тетрафенилэтан получен только симметрический (C6H5)2CH—CH(C6H5)2, темп. пл. 209°, тем. кип. 379—383°; образуется он легче всего при нагревании под давлением тиобензофенона (C6H5)2CS с порошкообразной медью.Тетрафенилэтилен (C6H5)2С=С(C6H5)2, темп. пл. 221°, получается из дифенилдихлорметана (C6H5)2CCl2 при действии на него металлического серебра.Все вышеописанные аром. У. характеризуются тем, что в них водороды бензольного кольца замещаются одноатомными радикалами, но существуют и такие производные бензола, в которых боковая цепь двухатомна и замещает сразу два атома водорода ядра, образуя с атомами углерода последнего новое кольцо. Из таких производных имеют наибольшее значение аром. У. с непредельной боковой цепью, смыкающейся при помощи рядом стоящих атомов углерода кольца. Теоретически в этом случае можно представить следующие типы подобных соединений:
и наконец этот последний хлорюр дает нормально толан:C6H5—CCl=CH—C6H5 — HCl = C6H5С=CC6H5[Очевидно, совершенно такое же толкование реакции можно применить и для случая образования стильбена из монохлордифенилэтана.].В ходе этих реакций в сторону образования симметрических соединений наблюдается некоторая аналогия с образованием симметрических ацетиленовых углеводородов жирного ряда из их несимметрических изомеров под влиянием едкого натра при высокой температуре. Реакцию эту изучал Фаворский и показал, что здесь всегда идет изомеризация в сторону образования наиболее симметрического продукта; то же самое мы наблюдаем и в вышеприведенных случаях, причем конечным результатом и здесь являются симметричный дифенилацетилен или дифенилэтилен. Ввиду такой аналогии возможно предположить вообще в производных этилена и ацетилена стремление образовать наиболее симметричные продукты, и в этом случае было бы интересно попробовать изомеризовать as-дифенилэтилен и дифенилацетилен в условиях реакции Фаворского. s-Дифенилэтан, или дибензил С6Н5—СН2—СН2—С6Н5, темп. пл. 52°, темп. кип. 284, получается обыкновенными синтетическими реакциями из C6Н5CH2Cl или восстановлением йодистоводородной кисл. бензоина C6H5CH(OH)—CO—C6H5, который, в свою очередь, легко образуется конденсацией бензойного альдегида в присутствии цианистого калия:C6H5CHO + OCH—C6H5 = C6H5CH(OH)—COC6H5.При нагревании до 500° дибензил претерпевает характерное разложение с выделением водорода, и в этом случае образуются стильбен, толан и фенантрен (см. ниже); при окислении же дибензил распадается, давая бензойную кислоту.s-Дифенилэтилен, или стильбен C6H5CH=CHC6H5, темп. пл. 124°, темп. кип. 306°, получается, кроме вышеуказанной реакции разложения дибензила и обыкновенных синтетических реакций, еще при многих других превращениях различных ароматических соединений, и потому он принадлежит к весьма давно открытым аром. У. В первый раз его получил Лоран в 1844 г. Между прочим, интересно его образование из дифенильного эфира фумаровой кислоты C6H5OCOCH=CH—COOC6H5, который при перегонке распадается на углекислоту и стильбен:C6H5OCO—CH=CH—COOC6H5 = 2CO2 + C6H5CH=CH—C6H5.Этот У., подобно этилену, способен присоединять 2 атома брома и давать s-дифенилдибромэтан C6H5Br—CHBr—C6H5, который, в свою очередь, при обмыливании спиртовым раствором едкого кали дает дифенилацетилен, или толан, C6H5C≡CC6H5, темп. пл. 60°.Трифенилэтан (C6H5)2CH—CH2C6H5 до сих пор не получен; но известны некоторые его производные.Тетрафенилэтан получен только симметрический (C6H5)2CH—CH(C6H5)2, темп. пл. 209°, тем. кип. 379—383°; образуется он легче всего при нагревании под давлением тиобензофенона (C6H5)2CS с порошкообразной медью.Тетрафенилэтилен (C6H5)2С=С(C6H5)2, темп. пл. 221°, получается из дифенилдихлорметана (C6H5)2CCl2 при действии на него металлического серебра.Все вышеописанные аром. У. характеризуются тем, что в них водороды бензольного кольца замещаются одноатомными радикалами, но существуют и такие производные бензола, в которых боковая цепь двухатомна и замещает сразу два атома водорода ядра, образуя с атомами углерода последнего новое кольцо. Из таких производных имеют наибольшее значение аром. У. с непредельной боковой цепью, смыкающейся при помощи рядом стоящих атомов углерода кольца. Теоретически в этом случае можно представить следующие типы подобных соединений: С другой стороны, возможно подобным же образом исходить из дифенила, и тогда получатся:
С другой стороны, возможно подобным же образом исходить из дифенила, и тогда получатся: Все эти аром. У. известны, кроме первых двух, и по своему характеру занимают середину между бензолом и его гомологами с открытыми цепями, так как в них новообразованное кольцо всегда в большей или меньшей мере приближается по свойствам к бензольному, в случае же, если оно вместе с замыкающими его атомами углерода бензольного кольца состоит из шести атомов (нафталин, антрацен, фенантрен и т. д.), то такое кольцо становится совершенно подобно бензольному, и потому нафталин можно рассматривать как продукт конденсации 2-х частиц, фенантрен — 3 частиц бензола и пр., и, следовательно, все то, что было сказано о бензоле и его производных, целиком относится и к подобным многокольчатым аром. У. Они так же, напр., относятся к азотной и серной кислотам, так же способны переходить в жирные кольчатые У., фиксируя водород в момент выделения, и подобно же бензолу и при аналогичных же условиях способны конденсироваться с жирными или ароматич. радикалами, образуя еще более сложные аром. У. Так, известны, напр., метилнафталин С10Н7—СН3, метилантрацен С14Н9—СН3 и др. Конденсируясь с бензолом, нафталин дает фенилнафталин С10Н7—C6Н5, наконец, две частицы нафталина образуют динафтил С10Н7—С10Н7, подобно тому как две частицы бензола при краснокалильном жаре уплотняются в дифенил. В свою очередь, исходя из последних двух У., возможно образовать очень сложные производные с замкнутой группировкой, подобно тому, как были образованы флуорен и фенантрен из дифенила. В этом случае из β-фенилнафталина получаются:
Все эти аром. У. известны, кроме первых двух, и по своему характеру занимают середину между бензолом и его гомологами с открытыми цепями, так как в них новообразованное кольцо всегда в большей или меньшей мере приближается по свойствам к бензольному, в случае же, если оно вместе с замыкающими его атомами углерода бензольного кольца состоит из шести атомов (нафталин, антрацен, фенантрен и т. д.), то такое кольцо становится совершенно подобно бензольному, и потому нафталин можно рассматривать как продукт конденсации 2-х частиц, фенантрен — 3 частиц бензола и пр., и, следовательно, все то, что было сказано о бензоле и его производных, целиком относится и к подобным многокольчатым аром. У. Они так же, напр., относятся к азотной и серной кислотам, так же способны переходить в жирные кольчатые У., фиксируя водород в момент выделения, и подобно же бензолу и при аналогичных же условиях способны конденсироваться с жирными или ароматич. радикалами, образуя еще более сложные аром. У. Так, известны, напр., метилнафталин С10Н7—СН3, метилантрацен С14Н9—СН3 и др. Конденсируясь с бензолом, нафталин дает фенилнафталин С10Н7—C6Н5, наконец, две частицы нафталина образуют динафтил С10Н7—С10Н7, подобно тому как две частицы бензола при краснокалильном жаре уплотняются в дифенил. В свою очередь, исходя из последних двух У., возможно образовать очень сложные производные с замкнутой группировкой, подобно тому, как были образованы флуорен и фенантрен из дифенила. В этом случае из β-фенилнафталина получаются: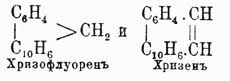 а из β-динафтила
а из β-динафтила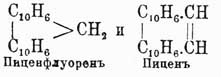 Далее флуорен может быть конденсирован сам с собой в бидифениленэтен
Далее флуорен может быть конденсирован сам с собой в бидифениленэтен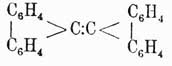 или с этиленным остатком в флуорантен, или идрил, строение которого выражается формулой
или с этиленным остатком в флуорантен, или идрил, строение которого выражается формулой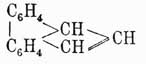 Из этого перечня видно, какое громадное разнообразие строений встречается у подобных сложных аром. У. Доказать его часто представляется делом великой трудности, и немудрено поэтому, что еще до сих пор имеются подобные сложные У., рациональная формула которых еще совершенно не определена. На практике, однако, значение имеют только нафталин, антрацен и отчасти фенантрен и флуорен; остальные же представляют или только научный интерес, или же интересны потому, что находятся в готовом виде в очень высококипящих фракциях каменноугольной и других смол.Инден
Из этого перечня видно, какое громадное разнообразие строений встречается у подобных сложных аром. У. Доказать его часто представляется делом великой трудности, и немудрено поэтому, что еще до сих пор имеются подобные сложные У., рациональная формула которых еще совершенно не определена. На практике, однако, значение имеют только нафталин, антрацен и отчасти фенантрен и флуорен; остальные же представляют или только научный интерес, или же интересны потому, что находятся в готовом виде в очень высококипящих фракциях каменноугольной и других смол.Инден , температура кипения 178°, находится вместе с кумароном
, температура кипения 178°, находится вместе с кумароном  , к которому он близок по своим свойствам, в фракции каменноугольной смолы, кипящей при 176—182°. Для его выделения из этой фракции определяют предварительно при помощи титрования бромом, сколько в ней находится непредельных соединений (индена и кумарона), и затем прибавляют туда необходимое для их насыщения количество пикриновой кислоты, рассчитывая, что 1 мол. последней соединяется с 1 мол. первых двух; смесь нагревают до растворения пикриновой кисл., и тогда по охлаждении жидкости выделяются кристаллы пикрата индена, который подвергают перегонке с водяным паром, при чем он распадается на инден, отгоняющийся с паром, и пикриновую кислоту, остающуюся в перегонном аппарате. Полученный сырой инден растворяют в толуоле и снова осаждают в виде пикрата, который опять вторично подвергается перегонке с водяным паром. Чистый инден представляет маслянистую бесцветную жидкость, легко окисляющуюся азотной кислотой во фталевую кислоту. Он присоединяет по месту двойной связи пятиатомного кольца 2 атома галоида, водородом в момент выделения превращается в гидринден
, к которому он близок по своим свойствам, в фракции каменноугольной смолы, кипящей при 176—182°. Для его выделения из этой фракции определяют предварительно при помощи титрования бромом, сколько в ней находится непредельных соединений (индена и кумарона), и затем прибавляют туда необходимое для их насыщения количество пикриновой кислоты, рассчитывая, что 1 мол. последней соединяется с 1 мол. первых двух; смесь нагревают до растворения пикриновой кисл., и тогда по охлаждении жидкости выделяются кристаллы пикрата индена, который подвергают перегонке с водяным паром, при чем он распадается на инден, отгоняющийся с паром, и пикриновую кислоту, остающуюся в перегонном аппарате. Полученный сырой инден растворяют в толуоле и снова осаждают в виде пикрата, который опять вторично подвергается перегонке с водяным паром. Чистый инден представляет маслянистую бесцветную жидкость, легко окисляющуюся азотной кислотой во фталевую кислоту. Он присоединяет по месту двойной связи пятиатомного кольца 2 атома галоида, водородом в момент выделения превращается в гидринден  , который кипит при той же самой температуре, как и инден. При краснокалильном жаре инден теряет из 2 частиц 4Н и переходит в хризен.Нафталин C10H8 был открыт Гарденом в продуктах перегонки каменноугольной смолы. Благодаря поразительному сходству по своим свойствам с бензолом вопрос о строении этого У. весьма интересовал химиков; но только в 1866 г. Гребе весьма остроумно доказал, что он имеет вышеприведенную рациональную формулу, данную ему впервые Эрленмейером [Доказательство строения нафталина, данное Гребе, приведено выше — при доказательстве строения фталевой кислоты.]. Впоследствии эта формула подтвердилась еще многими синтезами нафталина и различными продуктами распада его молекулы. Из этих реакций наиболее характерными являются: образование нафтола при нагревании фенилизокротоновой кисл.:
, который кипит при той же самой температуре, как и инден. При краснокалильном жаре инден теряет из 2 частиц 4Н и переходит в хризен.Нафталин C10H8 был открыт Гарденом в продуктах перегонки каменноугольной смолы. Благодаря поразительному сходству по своим свойствам с бензолом вопрос о строении этого У. весьма интересовал химиков; но только в 1866 г. Гребе весьма остроумно доказал, что он имеет вышеприведенную рациональную формулу, данную ему впервые Эрленмейером [Доказательство строения нафталина, данное Гребе, приведено выше — при доказательстве строения фталевой кислоты.]. Впоследствии эта формула подтвердилась еще многими синтезами нафталина и различными продуктами распада его молекулы. Из этих реакций наиболее характерными являются: образование нафтола при нагревании фенилизокротоновой кисл.: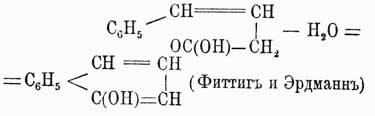 конденсация бромистого ксилилена с натриевой солью ацетилентетракарбонового эфира:
конденсация бромистого ксилилена с натриевой солью ацетилентетракарбонового эфира: (Байер и Перкин); полученный эфир при обмыливании дает тетрагидронафталиндикарбоновую кислоту, серебряная соль которой при перегонке разлагается на CO2, Н2 и нафталин. При окислении нафталина марганцово-калиевой солью между прочими продуктами образуется фенилглиоксилоортодикарбоновая кисл.
(Байер и Перкин); полученный эфир при обмыливании дает тетрагидронафталиндикарбоновую кислоту, серебряная соль которой при перегонке разлагается на CO2, Н2 и нафталин. При окислении нафталина марганцово-калиевой солью между прочими продуктами образуется фенилглиоксилоортодикарбоновая кисл. , а при подобном же окислении β-нафтола получается ортокоричнокарбоновая кисл.
, а при подобном же окислении β-нафтола получается ортокоричнокарбоновая кисл. Все это и много других еще данных с полной достоверностью устанавливают строение нафталина. Если теперь посмотреть на его формулу, то становится очевидным, что явления изомерии при замещении водородов нафталина будут значительно сложнее, чем при бензоле, так как в нем имеются два углеродных атома (не обозначенные греческими буквами), единицы сродства которых насыщены другими углеродными атомами и которые, следовательно, не способны присоединять боковых цепей, и затем карбинные группы (СН) разделяются на два различных класса, по четыре в каждом: одни из них (α1, α2, α3, α4) связаны с вышеуказанными двумя углеродами, а другие (β1....β4) занимают положение, аналогичное карбинным группам бензольного ядра.
Все это и много других еще данных с полной достоверностью устанавливают строение нафталина. Если теперь посмотреть на его формулу, то становится очевидным, что явления изомерии при замещении водородов нафталина будут значительно сложнее, чем при бензоле, так как в нем имеются два углеродных атома (не обозначенные греческими буквами), единицы сродства которых насыщены другими углеродными атомами и которые, следовательно, не способны присоединять боковых цепей, и затем карбинные группы (СН) разделяются на два различных класса, по четыре в каждом: одни из них (α1, α2, α3, α4) связаны с вышеуказанными двумя углеродами, а другие (β1....β4) занимают положение, аналогичное карбинным группам бензольного ядра.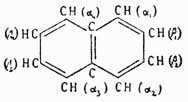 Поэтому в нафталине, следовательно, каждой карбинной группе отвечают только три ей тождественных. Тождество этих групп было доказано экспериментально Либерманном и Аттербергом, подобно тому, как это сделал Ладенбург для бензола. Из этого рассуждения вытекает, что даже монопроизводные нафталина должны иметься в двух изомерах — α- и β-ряда; двузамещенных должно быть 10 и т. д. Определение строения этих производных обыкновенно начинают с окисления, при чем одно из ядер нафталина сгорает до двух карбоксилов, связанных в ортоположении со вторым бензольным кольцом; следовательно, в этом случае всегда получатся производные фталевой кислоты, и по числу боковых групп в них можно судить, имело ли исследуемое производное нафталина боковые группы в одном или двух ядрах. Затем уже устанавливается самое положение боковых групп, опять-таки стараясь перевести исследуемое вещество в то или другое бензольное производное. В позднейшее время Бамбергер и Армстронг по аналогии с изменением строения бензольного кольца (см. выше) предложили и для нафталина формулу строения со свободными единицами сродства углеродов, направлеными в центры колец. Ввиду того, что нафталин вполне подобен по своим свойствам бензолу, и предложенные указанными учеными формулы для него имеют такое же точно основание, как и для бензола, так как здесь мы опять-таки сталкиваемся или с явлениями изомерии, или же с измененным характером непредельных соединений.Нафталин тверд; он плавится при 79° и кипит при 218°; получают его кристаллизацией при охлаждении из фракции каменноугольной смолы, кипящей при 180—300°. Такие широкие пределы этой фракции объясняются тем, что нафталин весьма летуч и легко перегоняется с парами низкокипящих У. Ввиду этой же летучести он обладает очень сильным запахом. Он способен соединяться с пикриновой кисл., давая кристаллич. продукт C10H8—C6H2(NO2)3—OH с темп. пл. 149°. При окислении дает фталевую кислоту, а при восстановлении (Na + амиловый спирт) переходит в гидронафталины, подобно тому, как бензол дает гидробензолы. При бензоле было указано, что эти гидросоединения обладают уже совершенно характером жирных У., гидронафталины же приобретают этот характер, только когда гидрогенизация распространяется на оба кольца. Нафталин употребляется как дезинфекционное средство, но главное свое значение приобрел в производстве красок, так как здесь он во многих случаях с успехом заменяет дорогостоящий бензол. Открытие нафталиновых пигментов произвело целый переворот в технике, так как до этого времени можно было опасаться, что заводы в силу недостатка бензоловых У. не в состоянии будут удовлетворить спрос покупателей органических красок, и в это же время нафталин, необходимо получающийся на заводах как побочный продукт, ложился только обременяющим балластом, почти не имевшим рыночной стоимости. В настоящее же время значительно более 50% всех органических красок содержат в своем составе этот У., в громадном большинстве случаев образующий пигменты несравненно лучшие, чем соответствующие бензоловые. Гомологи нафталина получаются совершенно так же, как гомологи бензола, но значения особенного они не имеют. В технике же играют роль его амидо- и оксипроизводные, или нафтиламины (см.) и нафтолы (см.), и их сульфоновые кислоты.Антрацен С14Н10 находится во фракции каменноугольной смолы, кипящей при 340—360°. Строение его легко устанавливается на основании следующих синтетических реакций:
Поэтому в нафталине, следовательно, каждой карбинной группе отвечают только три ей тождественных. Тождество этих групп было доказано экспериментально Либерманном и Аттербергом, подобно тому, как это сделал Ладенбург для бензола. Из этого рассуждения вытекает, что даже монопроизводные нафталина должны иметься в двух изомерах — α- и β-ряда; двузамещенных должно быть 10 и т. д. Определение строения этих производных обыкновенно начинают с окисления, при чем одно из ядер нафталина сгорает до двух карбоксилов, связанных в ортоположении со вторым бензольным кольцом; следовательно, в этом случае всегда получатся производные фталевой кислоты, и по числу боковых групп в них можно судить, имело ли исследуемое производное нафталина боковые группы в одном или двух ядрах. Затем уже устанавливается самое положение боковых групп, опять-таки стараясь перевести исследуемое вещество в то или другое бензольное производное. В позднейшее время Бамбергер и Армстронг по аналогии с изменением строения бензольного кольца (см. выше) предложили и для нафталина формулу строения со свободными единицами сродства углеродов, направлеными в центры колец. Ввиду того, что нафталин вполне подобен по своим свойствам бензолу, и предложенные указанными учеными формулы для него имеют такое же точно основание, как и для бензола, так как здесь мы опять-таки сталкиваемся или с явлениями изомерии, или же с измененным характером непредельных соединений.Нафталин тверд; он плавится при 79° и кипит при 218°; получают его кристаллизацией при охлаждении из фракции каменноугольной смолы, кипящей при 180—300°. Такие широкие пределы этой фракции объясняются тем, что нафталин весьма летуч и легко перегоняется с парами низкокипящих У. Ввиду этой же летучести он обладает очень сильным запахом. Он способен соединяться с пикриновой кисл., давая кристаллич. продукт C10H8—C6H2(NO2)3—OH с темп. пл. 149°. При окислении дает фталевую кислоту, а при восстановлении (Na + амиловый спирт) переходит в гидронафталины, подобно тому, как бензол дает гидробензолы. При бензоле было указано, что эти гидросоединения обладают уже совершенно характером жирных У., гидронафталины же приобретают этот характер, только когда гидрогенизация распространяется на оба кольца. Нафталин употребляется как дезинфекционное средство, но главное свое значение приобрел в производстве красок, так как здесь он во многих случаях с успехом заменяет дорогостоящий бензол. Открытие нафталиновых пигментов произвело целый переворот в технике, так как до этого времени можно было опасаться, что заводы в силу недостатка бензоловых У. не в состоянии будут удовлетворить спрос покупателей органических красок, и в это же время нафталин, необходимо получающийся на заводах как побочный продукт, ложился только обременяющим балластом, почти не имевшим рыночной стоимости. В настоящее же время значительно более 50% всех органических красок содержат в своем составе этот У., в громадном большинстве случаев образующий пигменты несравненно лучшие, чем соответствующие бензоловые. Гомологи нафталина получаются совершенно так же, как гомологи бензола, но значения особенного они не имеют. В технике же играют роль его амидо- и оксипроизводные, или нафтиламины (см.) и нафтолы (см.), и их сульфоновые кислоты.Антрацен С14Н10 находится во фракции каменноугольной смолы, кипящей при 340—360°. Строение его легко устанавливается на основании следующих синтетических реакций: В чистом виде антрацен получается из сырого продукта обработкой последнего жидким сернистым ангидридом, растворяющим все примеси и оставляющим в нерастворенном виде чистый продукт в форме белой кристаллической массы, плавящейся при 213° и кипящей при 315°. Он обладает замечательным свойством в растворе в ксилоле на солнечном свете полимеризоваться в парантрацен (C14H10)2, плавящийся при 244° и переходящий при этом в обыкновенное видоизменение. Эта способность полимеризации не наблюдается ни у нафталина, ни у бензола, и, вероятно, ее нужно приписать присутствию связи между средними карбинными группами. Вообще говоря, антрацен по своим свойствам весьма напоминает бензол и нафталин, хотя средние карбинные группы носят несколько иной характер, и в то время как производные типа
В чистом виде антрацен получается из сырого продукта обработкой последнего жидким сернистым ангидридом, растворяющим все примеси и оставляющим в нерастворенном виде чистый продукт в форме белой кристаллической массы, плавящейся при 213° и кипящей при 315°. Он обладает замечательным свойством в растворе в ксилоле на солнечном свете полимеризоваться в парантрацен (C14H10)2, плавящийся при 244° и переходящий при этом в обыкновенное видоизменение. Эта способность полимеризации не наблюдается ни у нафталина, ни у бензола, и, вероятно, ее нужно приписать присутствию связи между средними карбинными группами. Вообще говоря, антрацен по своим свойствам весьма напоминает бензол и нафталин, хотя средние карбинные группы носят несколько иной характер, и в то время как производные типа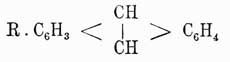 очень похожи на производные бензола; производные типа
очень похожи на производные бензола; производные типа скорее носят характер жирных соединений, связанных с бензольными ядрами. Например,
скорее носят характер жирных соединений, связанных с бензольными ядрами. Например, истинный фенол,а
истинный фенол,а третичный спирт.При окислении антрацен образует антрахинон
третичный спирт.При окислении антрацен образует антрахинон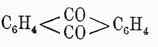 (см. Хиноны),оксипроизводные которого очень распространены в растительном царстве (см. Оксиантрахиноны) и некоторые из них представляют очень важные краски.Флуорен
(см. Хиноны),оксипроизводные которого очень распространены в растительном царстве (см. Оксиантрахиноны) и некоторые из них представляют очень важные краски.Флуорен , темп. пл. 113°, темп. кип. 295°, находится в каменноугольной смоле, синтетически может быть получен из ортодифеновой кислоты
, темп. пл. 113°, темп. кип. 295°, находится в каменноугольной смоле, синтетически может быть получен из ортодифеновой кислоты  , кальциевая соль которой при сухой перегонке дает дифениленкетон
, кальциевая соль которой при сухой перегонке дает дифениленкетон  , восстановляющийся при перегонке с цинковой пылью в флуорен. У. этот интересен тем, что при нагревании с окисью свинца он конденсируется, теряя 4 ат. водорода и переходя в бидифениленэтен
, восстановляющийся при перегонке с цинковой пылью в флуорен. У. этот интересен тем, что при нагревании с окисью свинца он конденсируется, теряя 4 ат. водорода и переходя в бидифениленэтен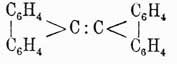 темп. пл. 188°, один из очень немногих У. (см.), окрашенных в ярко-красный цвет. При окислении флуорен дает дифениленкетон.Фенантрен
темп. пл. 188°, один из очень немногих У. (см.), окрашенных в ярко-красный цвет. При окислении флуорен дает дифениленкетон.Фенантрен изомерен антрацену, но, подобно нафталину, состоит как бы только из конденсированных бензольных ядер. Темп. пл. 99°, темп. кип. 340°, сопровождает антрацен в естественных продуктах. Синтетически легко получается пропусканием дибензила через раскаленные трубки:
изомерен антрацену, но, подобно нафталину, состоит как бы только из конденсированных бензольных ядер. Темп. пл. 99°, темп. кип. 340°, сопровождает антрацен в естественных продуктах. Синтетически легко получается пропусканием дибензила через раскаленные трубки: При окислении фенантрен дает сначала фенантренхинон
При окислении фенантрен дает сначала фенантренхинон , а затем дифеновую кислоту (см. выше). Его гомолог метилизопропилфенантрен, или ретен
, а затем дифеновую кислоту (см. выше). Его гомолог метилизопропилфенантрен, или ретен темп. пл. 98°, темп. кип. 394°, находится в смоле очень многих хвойных деревьев и в некоторых ископаемых смолах. Вообще производные фенантрена встречаются в продуктах растительного царства, и, например, алкалоид морфин, вероятно, принадлежит к этому классу органических соединений. В технике фенантрен до сих пор значения не имеет и, подобно нафталину в былые времена, представляет балласт для заводов.Пирен
темп. пл. 98°, темп. кип. 394°, находится в смоле очень многих хвойных деревьев и в некоторых ископаемых смолах. Вообще производные фенантрена встречаются в продуктах растительного царства, и, например, алкалоид морфин, вероятно, принадлежит к этому классу органических соединений. В технике фенантрен до сих пор значения не имеет и, подобно нафталину в былые времена, представляет балласт для заводов.Пирен , темп. пл. 148°, темп. кип. 260° при давлении в 60 мм, получается из наиболее высококипящих фракций каменноугольной смолы, где он находится вместе с флуорантеном, или идрилом, C15H10 (см. выше), темп. пл. 110°, темп. кип. 250° под давлением 60 мм. Оба эти У. образуют земляное сало в сланцевых залежах Идрии. Строение их еще не вполне установлено, но по своим свойствам они весьма близки к фенантрену.Хризен
, темп. пл. 148°, темп. кип. 260° при давлении в 60 мм, получается из наиболее высококипящих фракций каменноугольной смолы, где он находится вместе с флуорантеном, или идрилом, C15H10 (см. выше), темп. пл. 110°, темп. кип. 250° под давлением 60 мм. Оба эти У. образуют земляное сало в сланцевых залежах Идрии. Строение их еще не вполне установлено, но по своим свойствам они весьма близки к фенантрену.Хризен , темп. пл. 250°, темп. кип. 448°, строение имеет совершенно такое же, как фенантрен, только в нем вместо второго бензольного кольца имеется нафталиновое, подобно тому как пицен
, темп. пл. 250°, темп. кип. 448°, строение имеет совершенно такое же, как фенантрен, только в нем вместо второго бензольного кольца имеется нафталиновое, подобно тому как пицен  имеет оба кольца нафталиновые. Оба У. до крайности похожи на фенантрен и могут быть рассматриваемы: первый — как нафтофенантрен, а второй — как динафтофенантрен. Пицен представляет наиболее высокоплавящийся У. (темп. пл. 364°). Находится в неперегоняющихся остатках нефти и смолы бурых каменных углей.Д. А. Хардин. Δ.
имеет оба кольца нафталиновые. Оба У. до крайности похожи на фенантрен и могут быть рассматриваемы: первый — как нафтофенантрен, а второй — как динафтофенантрен. Пицен представляет наиболее высокоплавящийся У. (темп. пл. 364°). Находится в неперегоняющихся остатках нефти и смолы бурых каменных углей.Д. А. Хардин. Δ.
Энциклопедический словарь Ф.А. Брокгауза и И.А. Ефрона. — С.-Пб.: Брокгауз-Ефрон. 1890—1907.