- ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ КИНЕТИКА
изучает закономерности протекания во времени ферментативных р-ций, а также их механизм; раздел кинетики химической.
Каталитич. цикл конверсии в-ва S (субстрата) в продукт P под действием фермента E протекает с образованием промежут. соед. Xi:

где ki - константы скорости отдельных элементарных стадий,
При данной т-ре скорость р-ции зависит от концентраций фермента, субстрата и состава среды. Различают стационарную, предстационарную и релаксационную кинетику ферментативных р-ций.
Стационарная кинетика. В стационарном состоянии по промежуточным соед. (dXi/dt = 0, i = 1, ..., n) и при избытке субстрата
 , где [S]0 и [E]0 - начальные концентрации соотв. субстрата и фермента, кинетика процесса характеризуется постоянным, неизменным во времени уровнем концентраций промежут. соед., а выражение для скорости процесса v0, наз. начальной стационарной скоростью, имеет вид (ур-ние Михаэлиса- Ментен):
, где [S]0 и [E]0 - начальные концентрации соотв. субстрата и фермента, кинетика процесса характеризуется постоянным, неизменным во времени уровнем концентраций промежут. соед., а выражение для скорости процесса v0, наз. начальной стационарной скоростью, имеет вид (ур-ние Михаэлиса- Ментен):
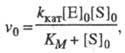 (1)
(1)
где значения k кат и К м -> ф-ции констант скорости элементарных стадий и заданы ур-нениями:
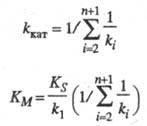
Величину k кат наз. эффективной каталитич. константой скорости процесса, параметр К м -> константой Михаэлиса. Значение k кат определяется величинами
При больших концентрациях субстрата, когда
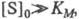
 т. е. скорость р-ции не зависит от концентрации субстрата и достигает постоянной величины, наз. макс. скоростью. Графически ур-ние Михаэлиса - Ментен представляет собой гиперболу. Его можно линеаризовать, используя метод двойных обратных величин (метод Лайнуи-вера - Берка), т. е. строя зависимость 1/vот 1/[S]0, или др. методы. Линейная форма ур-ния (1) имеет вид:
т. е. скорость р-ции не зависит от концентрации субстрата и достигает постоянной величины, наз. макс. скоростью. Графически ур-ние Михаэлиса - Ментен представляет собой гиперболу. Его можно линеаризовать, используя метод двойных обратных величин (метод Лайнуи-вера - Берка), т. е. строя зависимость 1/vот 1/[S]0, или др. методы. Линейная форма ур-ния (1) имеет вид:
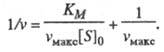 (2)
(2)
Она позволяет определить графически значения К м и v макс (рис. 1).
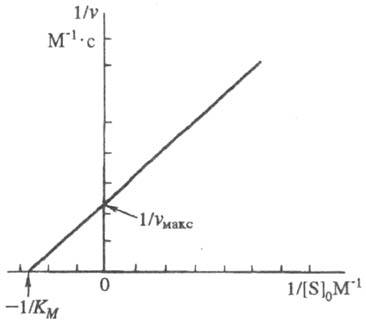
Рис. 1. График линейной трансформации ур-ния Михаэлиса - Ментен в двойных обратных величинах (по Лайнуиверу - Берку).
Величина К м > численно равна концентрации субстрата, при к-рой скорость р-ции равна
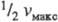 , поэтому К м часто служит мерой сродства субстрата и фермента, однако это справедливо лишь, если
, поэтому К м часто служит мерой сродства субстрата и фермента, однако это справедливо лишь, если 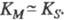
Величины К м > и
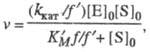
где f = 1 + <[H+]/K а + Kb/[H+] и f ' = 1 + <[H+]/К' а >+ K'b/>[H+] -т. наз. рН-ф-ции Михаэлиса, а К а, К b и К'a, K'b-> константы ионизации групп аи bсоотв. своб. фермента и фермент-субстратного комплекса. В координатах lg

Рис. 2. Зависимость каталитич. константы от рН в логарифмич. координатах.
Скорость ферментативной р-ции не всегда подчиняется ур-нию (1). Один из часто встречающихся случаев - участие в р-ции аллостерич. ферментов (см. Регуляторы ферментов), для к-рых зависимость степени насыщения фермента от [S]0 имеет негиперболич. характер (рис. 3). Это явление обусловлено кооперативностью связывания субстрата, т. е. когда связывание субстрата на одном из участков макромолекулы фермента увеличивает (положит. кооперативность) или уменьшает (отрицат. кооперативность) сродство к субстрату др. участка.
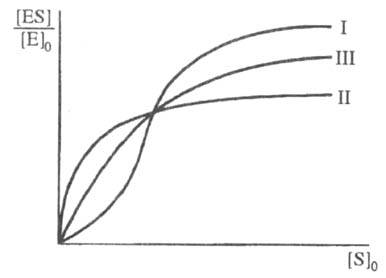
Рис. З Зависимость степени насыщения фермента субстратом от концентрации субстрата при положительной (I) и отрицательной (II) кооперативности, а также в ее отсутствии (III).
Предстационарная кинетика. При быстром смешении р-ров фермента и субстрата в интервале времен 10-6-10-1 с можно наблюдать переходные процессы, предшествующие образованию устойчивого стационарного состояния. В этом предстационарном режиме при использовании большого избытка субстрата
 система дифференц. ур-ний, описывающая кинетику процессов, линейна. Решение данного типа системы линейных дифференц. ур-ний дается суммой экспоненциальных членов. Так, для кинетич. схемы, представленной выше, кинетика накопления продукта имеет вид:
система дифференц. ур-ний, описывающая кинетику процессов, линейна. Решение данного типа системы линейных дифференц. ур-ний дается суммой экспоненциальных членов. Так, для кинетич. схемы, представленной выше, кинетика накопления продукта имеет вид:
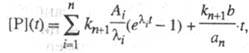
где Ai->, b, а n -> ф-ции элементарных констант скорости;
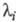 -корни соответствующего характеристич. ур-ния.
-корни соответствующего характеристич. ур-ния.
Величина, обратная
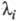 , наз. характеристич. временем процесса:
, наз. характеристич. временем процесса:
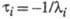
Для р-ции, протекающей с участием nпромежут. соед., можно получить nхарактеристич. времен.
Исследование кинетики ферментативной р-ции в предстационарном режиме позволяет получить представление о детальном механизме каталитич. цикла и определить константы скорости элементарных стадий процесса.
Экспериментально кинетику ферментативной р-ции в предстационарном режиме исследуют с помощью метода остановленной струи (см. Струевые кинетические методы), позволяющего смешивать компоненты р-ции в течение 1 мс.
Релаксационная кинетика. При быстром возмущающем воздействии на систему (изменение т-ры, давления, электрич. поля) время, к-рое необходимо системе для достижения нового равновесия или стационарного состояния, зависит от скорости процессов, определяющих каталитич. ферментативный цикл.
Система ур-ний, описывающая кинетику процесса, линейна, если смещение от положения равновесия невелико. Решение системы приводит к зависимостям концентраций компонентов разл. стадий процесса в виде суммы экспоненциальных членов, показатели экспонент к-рых имеют характер времен релаксаций. Результатом исследования является спектр времен релаксации, соответствующий числу промежут. соед., участвующих в процессе. Величины времен релаксаций зависят от констант скорости элементарных стадий процессов.
Релаксационные методы кинетики позволяют определить константы скорости отдельных элементарных стадий трансформации интермедиатов. Методы изучения релаксационной кинетики имеют разл. разрешающую способность: поглощение ультразвука - 10-6-10-10 с, температурный скачок - 1O-4-10-6 с, метод электрич. импульса - 10-4-10-6 с, скачок давления - 10-2 с. При исследовании кинетики ферментативных р-ций наиб, применение нашел метод температурного скачка.
Макрокинетика ферментативных процессов. Развитие методов получения гетерогенных катализаторов путем иммобилизации ферментов на разл. носителях (см. Иммобилизованные ферменты )обусловило необходимость анализа кинетики процессов с учетом массопереноса субстрата. Теоретически и экспериментально исследованы закономерности кинетики р-ций с учетом эффектов диффузионного слоя и для систем с внутридиффузионными затруднениями при распределении фермента внутри носителя.
В условиях, когда на кинетику процесса влияет диффузионный перенос субстрата, каталитич. эффективность системы уменьшается. Фактор эффективности
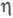 равен отношению плотности потока продукта в условиях протекания ферментативной р-ции с диффузионно пониженной концентрацией субстрата к потоку, к-рый мог бы реализоваться в отсутствие диффузионных ограничений. В чисто диффузионной области, когда скорость процесса определяется массопереносом субстрата, фактор эффективности для систем с внешнедиффузи-онным торможением обратно пропорционален диффузионному модулю
равен отношению плотности потока продукта в условиях протекания ферментативной р-ции с диффузионно пониженной концентрацией субстрата к потоку, к-рый мог бы реализоваться в отсутствие диффузионных ограничений. В чисто диффузионной области, когда скорость процесса определяется массопереносом субстрата, фактор эффективности для систем с внешнедиффузи-онным торможением обратно пропорционален диффузионному модулю  :
: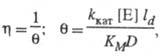
где
Для систем с внутридиффузионным торможением в р-циях первого порядка
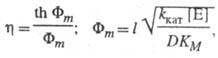
где Ф т - безразмерный модуль (модуль Тиле).
При анализе кинетич. закономерностей в ферментативных реакторах широкое теоретич. и эксперим. развитие получили "идеальные" модели реакторов, проточный безградиентный реактор (проточный реактор идеального перемешивания), проточный реактор с идеальным вытеснением, мембранный реактор.
Кинетика полиферментных процессов. В организме (клетке) ферменты действуют не изолированно, а катализируют цепи трансформации молекул. Р-ции в полиферментных системах с кинетич. точки зрения можно рассматривать как последоват. процессы, специфич. особенностью к-рых является катализ ферментами каждой из стадий:
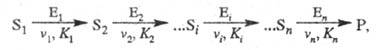
где
Важная особенность процесса - возможность образования устойчивого стационарного состояния. Условием-его возникновения может служить неравенство
Специфич. группу полиферментных систем составляют системы, осуществляющие окислит.-восстановит. р-ции с участием белковых переносчиков электронов. Переносчики образуют специфич. структуры, комплексы с детерминированной последовательностью переноса электрона. Кинетич. описание такого рода систем рассматривает в качестве независимой переменной состояния цепей с разл. степенью заселенности электронами.
Применение. Ф. р. к. широко используют в исследовательской практике для изучения механизмов действия ферментов и ферментных систем. Практически значимая область науки о ферментах - инженерная энзимология, оперирует понятиями Ф. р. к. для оптимизации биотехнол. процессов.
Лит.: Полторак О. M., Чухрай E. С, Физико-химические основы ферментативного катализа, M., 1971; Березин И. В., Мартинек К, Основы физической химии ферментативного катализа, M., 1977; Варфоломеев С. Д., Зайцев С. В., Кинетические методы в биохимических исследованиях, M.. 1982. С. Д. Варфоломеев.
Химическая энциклопедия. — М.: Советская энциклопедия. Под ред. И. Л. Кнунянца. 1988.