- Стереоизомерия
-
(хим.) — слово, употребляемое для обозначения изомерии (см.) таких соединений, отличие свойств которых объясняется различным пространственным (стереометрическим) расположением атомов в структурно тождественных молекулах. С. непосредственно связана с присутствием в молекулах соединений многовалентных (обладающих несколькими — тремя, четырьмя, пятью — единицами сродства) атомов. В настоящей статье будет более подробно затронута С. углеродных соединений и очень кратко — азотных. Историю вопроса — см. Стереохимия и Винная кислота. Между стереоизомерными соединениями надо отличать: 1) оптические изомеры, которыми являются тела, обладающие тождественными химическими и физическими свойствами, за исключением влияния на поляризованный луч, и изомерия которых обусловлена энантиоморфно асимметричным строением их молекул: известны углеродные и азотные оптические изомеры; к оптическим изомерам нужно отнести и рацемические тела; 2) так называемые геометрические изомеры, под которыми подразумеваются тела, не действующие на поляризованный луч, различие свойств которых (химических и физических), не объяснимое формулами на плоскости, объясняется пространственными формулами; сюда относятся: С. тел мезовинного типа; С. соединений, заключающих одну или несколько двойных связей между углеродными атомами; С. насыщенных циклических соединений; С. соединений, заключающих атомы углерода и азота, связанные многократными связями, и, наконец, геометрическая С. — азота.I) Оптические изомеры. Несмотря на громадное число работ, произведенных над оптически деятельными веществами со времени появления стереохимических воззрений (см.), мы почти ничего не можем прибавить нового к классической характеристике этих тел, сделанной Пастером при работе над винными кислотами. Всегда изомеру, вращающему плоскость поляризации прямолинейно поляризованного луча вправо, отвечает вполне тождественный по темп. плавления, темп. кипения, удельному весу, коэффициенту светопреломления и другим подобным физическим свойствам [Имеются несколько указаний на неравную растворимость некоторых оптических антиподов. Так, по Пьютти, d-аспарагин несколько растворимее l-аспарагина; по Юнгфлейшу, при охлаждении горячего раствора бромокамфорной кислоты первым выпадает l-изомер, и только ниже +40° — d-изомер. Возможно, что в обоих случаях противоположно вращающие изомеры не представляют настоящих оптических антиподов (Ландольф).] — изомер, который вращает плоскость поляризации влево и притом на одну и ту же абсолютную величину. Обыкновенно эти изомеры носят название оптических антиподов. Оптические антиподы вполне тождественны и по химическим свойствам. В этом современные воззрения расходятся с представлениями Пастера; последний, исходя из возможности деления правой и левой винной кислот (см.) посредством солей хинина, писал (1860): "хинин относится к обоим винным кислотам иначе, чем едкое кали, и потому иначе, что он асимметричен, а едкое кали — нет. Асимметрия является потому свойством, способным само по себе изменять химическое сродство". Наблюдение Пастера объясняется, однако, следующим образом. Оптические антиподы обладают тождественными физическими и химическими свойствами (в том числе и тождественной растворимостью), пока они геометрически энантиоморфны; когда же мы соединяем право- и левовращающую кислоту с оптически деятельным основанием, то мы образуем пару тел, которые уже не представляют зеркальных изображений друг друга (оптическая деятельность их различна), а потому они должны быть рассматриваемы просто как два изомерных тела, химические и физические свойства которых должны различаться; коэффициенты их растворимости будут различные, а, след., смесь таких изомеров может делиться только при фракционированной (ср. ниже) кристаллизации (Марквальд и Хволлес). Опыт действительно показывает, что если взята оптически недеятельная смесь правого и левого антиподов (напр., недеятельная метилэтилуксусная — см. Валерьяновая кисл., или недеятельная миндальная кисл. — см.) и нейтрализована наполовину в разбавленном водном растворе бруцином, то эфир извлекает оптически недеятельную же кислоту, что было бы невозможно при неравномерном распределении бруцина между оптическими антиподами. Повышение температуры кипения водных растворов солей цинхонина правой и левой винной кислот при одинаковой концентрации солей — тождественно, что указывает на одинаковую степень гидролиза (см.) этих солей и, наконец, скорость омыления метиловых эфиров правой и левой винных кислот никотином одна и та же (Марквальд и Хволлес). Наиболее обогатились наблюдениями последних лет наши сведения о рацемических соединениях (см.), и тут современные представления отличаются не только от представлений времен Пастера, но и являются гораздо более определенными, чем сообщенные под словом Рацемия (см.). Во-первых (благодаря главным образом теоретическому разбору явления Б. Роозебоомом с точки зрения правила фаз, см.), выработаны приемы, позволяющие вполне точно решить, представляет ли вещество, образуемое взаимодействием оптических антиподов, их смесь или же действительное соединение — рацемическое, т. е. типа виноградной кисл. (см.), тело кристаллическое, однородной структуры, содержащее равные количества энантиоморфных изомеров. Прием состоит в исследовании хода кривой температур плавления, иначе — хода взаимной растворимости оптических антиподов. Когда два реагирующих тела относятся друг к другу только как растворители (см.), то наблюдаются при исследовании их взаимной растворимости две кривых; каждая исходит из темп. плавл. соответственного тела; обе выражают собою понижение темп. плавл. растворителя под влиянием растворенного тела; обе пересекаются в эвтектической точке. При тождестве физических и химических свойств оптических антиподов ход обеих кривых должен быть тождественным; фигура должна быть, след., вполне симметричной и эвтектическая смесь отвечать составу: 50% d-изомера + 50% l-изомера (см.), что и изображено на фиг. 1, где по осям у-ов отложены темп., а по оси х-ов — процентное содержание антиподов в смеси;
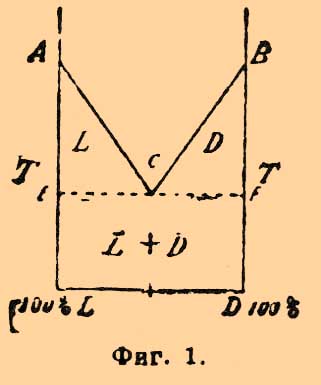 Фиг. 1.А и В представляют темп. плавл. исходных соединений; С — их эвтектическую точку; в треугольники BCF выпадает из сплава в твердом виде d-изомер, в треугольнике АСЕ — l-изомер; в области EFDL — возможна смесь кристаллов d- и l-изомеров, для которой Б. Роозебоомом предложено характерное название "конгломерата d+l". Прямолинейность линий АС и СВ схематическая: на самом деле, как видно, напр., из опытов Центнершвера над смесью d- и l-изомеров хлороянтарной кислоты Вальдена, они являются кривыми; кривые должны быть ломаными, если d- и l-изомеры наклонны к полиморфизму (Кипинг, Роозебоом). Когда реагирующие тела образуют соединение, способное плавиться без разложения, то предыдущая диаграмма как бы удваивается: темп. плавл. обоих реагирующих тел А и В может понижаться образующимся соединением АВ, а с другой стороны, и темп. плавл. АВ, лежащего по составу между А и B, может понижаться при растворении в нем как тела А, так и тела В; точка плавления AB должна лежать, таким образом, между двумя эвтектическими точками; она может быть притом выше и ниже точек плавления тел А и В. При перенесении этих соображений на оптические антиподы мы должны ожидать возможности двух вполне симметричных фигур (2 и 3); темп. плавл. инактивного (рацемического) соединения (точка E обеих диаграмм) должна отвечать и тут содержанию 50% d- и 50% l-изомеров.
Фиг. 1.А и В представляют темп. плавл. исходных соединений; С — их эвтектическую точку; в треугольники BCF выпадает из сплава в твердом виде d-изомер, в треугольнике АСЕ — l-изомер; в области EFDL — возможна смесь кристаллов d- и l-изомеров, для которой Б. Роозебоомом предложено характерное название "конгломерата d+l". Прямолинейность линий АС и СВ схематическая: на самом деле, как видно, напр., из опытов Центнершвера над смесью d- и l-изомеров хлороянтарной кислоты Вальдена, они являются кривыми; кривые должны быть ломаными, если d- и l-изомеры наклонны к полиморфизму (Кипинг, Роозебоом). Когда реагирующие тела образуют соединение, способное плавиться без разложения, то предыдущая диаграмма как бы удваивается: темп. плавл. обоих реагирующих тел А и В может понижаться образующимся соединением АВ, а с другой стороны, и темп. плавл. АВ, лежащего по составу между А и B, может понижаться при растворении в нем как тела А, так и тела В; точка плавления AB должна лежать, таким образом, между двумя эвтектическими точками; она может быть притом выше и ниже точек плавления тел А и В. При перенесении этих соображений на оптические антиподы мы должны ожидать возможности двух вполне симметричных фигур (2 и 3); темп. плавл. инактивного (рацемического) соединения (точка E обеих диаграмм) должна отвечать и тут содержанию 50% d- и 50% l-изомеров.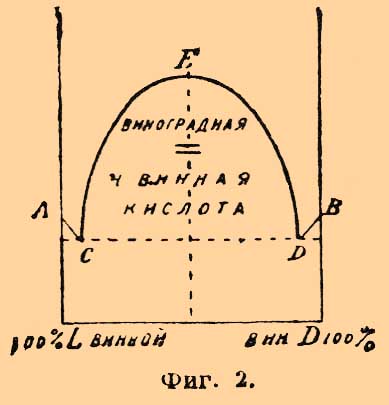 Фиг. 2.Фиг. 2 представляет явления, наблюдаемые для смесей правой (D) и левой (L) винных и виноградной (r-винной) кисл.; как видно, растворимость виноградной кисл. в винных очень незначительна; по-видимому, это явление довольно общее, так как подобное же отношение наблюдается для кислот d+l-камфарных и d+l-изокамфорных (Б. Роозебоом, Центнершвер [Центнершверу не удалось найти для винных кислот эвтектических точек С и D (фиг. 2).]. Фиг. 3 представляет в несколько преувеличенном виде явления [Диаграмма близко отвечает тому, что наблюдается для сплавов AgNO3 и TlNO3; обе соли плаватся почти при одной и той же температуре и дают соединение AgTl(NO3)2.], наблюдающиеся для кислот: d+l-бензиламиноянтарных и d+l-аминоянтарных (Центнершвер).
Фиг. 2.Фиг. 2 представляет явления, наблюдаемые для смесей правой (D) и левой (L) винных и виноградной (r-винной) кисл.; как видно, растворимость виноградной кисл. в винных очень незначительна; по-видимому, это явление довольно общее, так как подобное же отношение наблюдается для кислот d+l-камфарных и d+l-изокамфорных (Б. Роозебоом, Центнершвер [Центнершверу не удалось найти для винных кислот эвтектических точек С и D (фиг. 2).]. Фиг. 3 представляет в несколько преувеличенном виде явления [Диаграмма близко отвечает тому, что наблюдается для сплавов AgNO3 и TlNO3; обе соли плаватся почти при одной и той же температуре и дают соединение AgTl(NO3)2.], наблюдающиеся для кислот: d+l-бензиламиноянтарных и d+l-аминоянтарных (Центнершвер).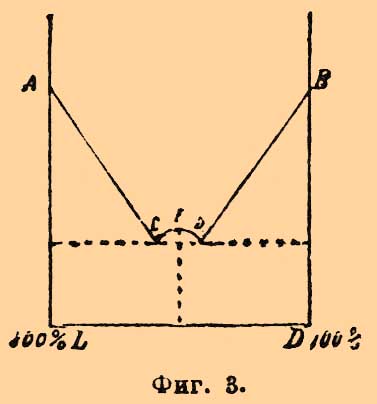 Фиг. 3Может быть, этому же типу отвечает и смесь d+l-хлороянтарных кислот (см. выше), по поводу которой Центнершвер замечает, что "темп. плавления инактивной смеси всегда отвечает относительной максимальной точке кривой, независимо от того, лежит ли эта температура выше или ниже температуры плавления оптически деятельных изомеров". Принимая во внимание, что удельный вес тех рацемических полимеров, темп. плавл. которых лежит выше темп. плавл. оптических антиподов, их составляющих, больше уд. веса этих последних (Вальден), меньше — в том случае, когда темп. плавл. рацемического вещества (50% d- + 50% l-изомеров) ниже темп. плавл. активных изомеров (Вальден), надо думать, что вполне аналогичные кривые получатся при нанесении по оси у-ов уд. весов, а по оси х-ов процентного состава изследуемых смесей. То же самое замечание приложимо к кривым растворимости их в воде (ср. Б. Роозебоом); впрочем, тут явление несколько усложняется распадением в разбавленных растворах рацемических соединений на оптические антиподы. Во-вторых — Ладенбургом (в сотрудничестве с Герцом и Доктором) доказана возможность существования в растворах частично рацемических соединений (partielle Racemie). Явление заключается в том, что некоторые средние соли рацемических кислот и оптически деятельных оснований способны существовать в растворах как таковые, несмотря на то, что оптическая деятельность солей правого и левого изомеров в этих случаях различная и, как указано выше, они не представляют энантиоморфных форм, а потому должны бы относиться друг к другу, как простые изомеры (стереоизомеры). Так, напр., средняя соль хинина и рацемической пировинной кислоты (метилянтарной, см. Янтарная кислота) плавится при 174°—175° и растворима (1 в. ч.) в 31,25 в. ч. спирта, между тем как соль хинина же и d-пировинной кисл. плавится при 169°—171° и растворима (1 в. ч.) в 23,80 в. ч. спирта, а соль хинина и l-пировинной кислоты гораздо растворимее в спирте [Нечистая, содержащая 74% l-соли, растворима 1 в. ч. в 6,66 в. ч. спирта.]. Затем — средняя соль стрихнина и виноградной кислоты (см. Винные кислоты) — 2С4H6О6(С21Н22N2O2)2+13Н2О — обладает уд. весом d20 = 1,4696, плавится при 222°, растворима (при 20°) в 2,45 в. ч. воды, а формула соли d-кисл. — C4H6O6(C21H22N2O2)+7H2O, ее уд. в. 1,5429, темп. пл. 228° и раствор. в 2,03 воды, формула же соли l-кислоты — С4H6О6(С21N2H22O2)2+3,5H2O (?), уд. в. 1,6080, темп. плавл. 242° и раствор. в 1,70 воды; подробное исследование показало, что соль 2С4H6О6(С21Н22N2O2)2+13Н2О способна существовать только до +29,5°; выше этой темп. она распадается на смесь солей d- и l-винных кислот (Ладенбург и Доктор). Что касается кривых плавления, то при частичной рацемии характер их тот же, что и при рацемии; вся разница в том, что температуры плавления исходных изомеров не тождественны. Еще несколько случаев частичной рацемии описано Попом (с Пичей и Гарвеем), Андреоччи, Вальденом и Центнершвером. Явление должно считать в высшей степени важным для практической стереохимии, так как неудача опытов активирования посредством взаимодействия данного инактивного соединения с активным телом не может отныне считаться абсолютным доказательством того, что исследуемое тело не принадлежит к рацемическим (Ладенбург). В-третьих, Поп с Кипингом установили возможность псевдорацемии. Явление наблюдается в том случае, когда кристаллографические свойства инактивной смеси (?) почти сливаются с кристаллографическими свойствами оптических антиподов; тогда возможно срастание равных (или приблизительно равных) весовых частей этих последних) с сохранением их кристаллической структуры; кристаллы псевдорацемических соединений — неоднородной структуры. — Исследование всей кривой плавления смесей d- и l-изомеров позволяет решить вопрос и о псевдорацемии. Кипинг и Поп высказали предположение, что любые смеси d- и l-изомеров должны в случае псевдорацемии плавиться при одной и той же температуре и именно температуре плавления взятых оптических антиподов. Этому случаю отвечает фиг. 4; тут имеется только одна линия плавления, каждый сплав застывает при постоянной темп., и состав жидкости одинаков с составом выпадающих из нее кристаллов.
Фиг. 3Может быть, этому же типу отвечает и смесь d+l-хлороянтарных кислот (см. выше), по поводу которой Центнершвер замечает, что "темп. плавления инактивной смеси всегда отвечает относительной максимальной точке кривой, независимо от того, лежит ли эта температура выше или ниже температуры плавления оптически деятельных изомеров". Принимая во внимание, что удельный вес тех рацемических полимеров, темп. плавл. которых лежит выше темп. плавл. оптических антиподов, их составляющих, больше уд. веса этих последних (Вальден), меньше — в том случае, когда темп. плавл. рацемического вещества (50% d- + 50% l-изомеров) ниже темп. плавл. активных изомеров (Вальден), надо думать, что вполне аналогичные кривые получатся при нанесении по оси у-ов уд. весов, а по оси х-ов процентного состава изследуемых смесей. То же самое замечание приложимо к кривым растворимости их в воде (ср. Б. Роозебоом); впрочем, тут явление несколько усложняется распадением в разбавленных растворах рацемических соединений на оптические антиподы. Во-вторых — Ладенбургом (в сотрудничестве с Герцом и Доктором) доказана возможность существования в растворах частично рацемических соединений (partielle Racemie). Явление заключается в том, что некоторые средние соли рацемических кислот и оптически деятельных оснований способны существовать в растворах как таковые, несмотря на то, что оптическая деятельность солей правого и левого изомеров в этих случаях различная и, как указано выше, они не представляют энантиоморфных форм, а потому должны бы относиться друг к другу, как простые изомеры (стереоизомеры). Так, напр., средняя соль хинина и рацемической пировинной кислоты (метилянтарной, см. Янтарная кислота) плавится при 174°—175° и растворима (1 в. ч.) в 31,25 в. ч. спирта, между тем как соль хинина же и d-пировинной кисл. плавится при 169°—171° и растворима (1 в. ч.) в 23,80 в. ч. спирта, а соль хинина и l-пировинной кислоты гораздо растворимее в спирте [Нечистая, содержащая 74% l-соли, растворима 1 в. ч. в 6,66 в. ч. спирта.]. Затем — средняя соль стрихнина и виноградной кислоты (см. Винные кислоты) — 2С4H6О6(С21Н22N2O2)2+13Н2О — обладает уд. весом d20 = 1,4696, плавится при 222°, растворима (при 20°) в 2,45 в. ч. воды, а формула соли d-кисл. — C4H6O6(C21H22N2O2)+7H2O, ее уд. в. 1,5429, темп. пл. 228° и раствор. в 2,03 воды, формула же соли l-кислоты — С4H6О6(С21N2H22O2)2+3,5H2O (?), уд. в. 1,6080, темп. плавл. 242° и раствор. в 1,70 воды; подробное исследование показало, что соль 2С4H6О6(С21Н22N2O2)2+13Н2О способна существовать только до +29,5°; выше этой темп. она распадается на смесь солей d- и l-винных кислот (Ладенбург и Доктор). Что касается кривых плавления, то при частичной рацемии характер их тот же, что и при рацемии; вся разница в том, что температуры плавления исходных изомеров не тождественны. Еще несколько случаев частичной рацемии описано Попом (с Пичей и Гарвеем), Андреоччи, Вальденом и Центнершвером. Явление должно считать в высшей степени важным для практической стереохимии, так как неудача опытов активирования посредством взаимодействия данного инактивного соединения с активным телом не может отныне считаться абсолютным доказательством того, что исследуемое тело не принадлежит к рацемическим (Ладенбург). В-третьих, Поп с Кипингом установили возможность псевдорацемии. Явление наблюдается в том случае, когда кристаллографические свойства инактивной смеси (?) почти сливаются с кристаллографическими свойствами оптических антиподов; тогда возможно срастание равных (или приблизительно равных) весовых частей этих последних) с сохранением их кристаллической структуры; кристаллы псевдорацемических соединений — неоднородной структуры. — Исследование всей кривой плавления смесей d- и l-изомеров позволяет решить вопрос и о псевдорацемии. Кипинг и Поп высказали предположение, что любые смеси d- и l-изомеров должны в случае псевдорацемии плавиться при одной и той же температуре и именно температуре плавления взятых оптических антиподов. Этому случаю отвечает фиг. 4; тут имеется только одна линия плавления, каждый сплав застывает при постоянной темп., и состав жидкости одинаков с составом выпадающих из нее кристаллов.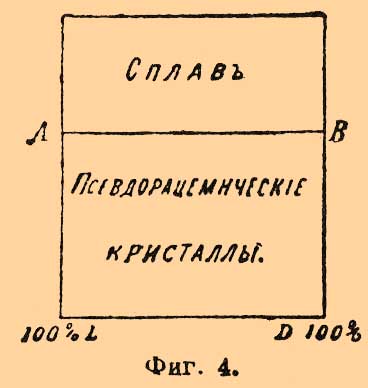 Фиг. 4По мнению Б. Роозебоома, такие случаи возможны, но только в виде исключений; наиболее же вероятные случаи должны отвечать схематическим диаграммам фиг. 5 и фиг. 6.
Фиг. 4По мнению Б. Роозебоома, такие случаи возможны, но только в виде исключений; наиболее же вероятные случаи должны отвечать схематическим диаграммам фиг. 5 и фиг. 6. Фиг. 5. Фиг. 6Верхняя кривая есть кривая температур застывания; нижняя — дает состав кристаллов, выпадающих в самом начале застывания из жидкого сплава; соответственные точки верхней и нижней кривых лежат на горизонтальной линии (ab — фиг. 5 и 6); отрезки ординат (ас — фиг. 5 и 6) между кривыми дают разность температур начала и конца застывания или начала и конца плавления. Благодаря симметрии как максимум, так и минимум приходятся на смеси: 50% d-изомера + 50% l-изомера; в этих точках верхняя и нижняя кривые на самом деле соприкасаются, а это значит, что инактивные кристаллические смеси плавятся при постоянной температуре. Таким образом, псевдорацемия характеризуется непрерывностью кривых плавления и застывания. К оптически деятельным веществам, способным к псевдорацемии типа фиг. 6, Б. Роозебоом относит исследованные Кипингом хлор- и бромангидриды сульфокамфорной кислоты и трибромкарбон из гидрокарбона. Чтобы закончить о рацемических соединениях, необходимо указать, что Поп, Пичей, Гарвей и Рич рекомендуют для разделения рацемических оснований d-α-хлоро- и бромокамфорсульфоновую кислоту — С10Н14ClO·SO3Н и C10H14BrO·SO3H; при помощи этих реактивов им удалось разделить: тетрагидропапаверин — С20Н25NO4, тетрагидрохиналдин — C10H13N, тетрагидропаратолухиналдин — C11H15N, тетрагидро-бета-нафтиламин С10Н12NH2 и камфороксим — С10Н15NOH. Затем Э. Фишер указал, что при активировании слабых амидокислот удобнее всего исходить из бензоилированных производных, которые более постоянны и легче кристаллизуются, чем амидокислоты, и из которых бензоильная группа может быть легко элиминирована. Этим путем с помощью бруцина и стрихнина ему удалось разделить — аланин, аспарагиновую и глутаминовую кислоты и тирозин. Наконец, Марквальд (с Маккензи) и Вальден показали возможность разделения рацемических соединений при взаимодействии их с оптически деятельными веществами на основании различной скорости реакции для d- и l-изомеров, которая возможна потому, что получаемые производные не являются оптически энантиоморфными. Первым удалось получить небольшое количество l-миндальной кислоты (см.) при неполной этерификации l-ментолом r-миндальной кислоты; Вальдену удалось констатировать незначительное разделение при этерификации l-амиловым спиртом бромангидрида r-бромопропионовой кислоты [Вполне подобные опыты Фрейндлера и Симона дали ранее отрицательные результаты.]. Как замечает Э. Фишер, способ по существу тождествен с так называемым биологическим методом разделения оптических антиподов Пастера, так как в последнем случае процесс сводится на взаимодействие рацемического вещества с оптически деятельной энзимой, вырабатываемой данным микроорганизмом.Все приведенные факты находятся в полной независимости от стереохимической гипотезы (см. Стереохимия) Фант-Гоффа и Ле-Беля; они не противоречат ей, но могли быть установлены даже на почве тех представлений об оптически деятельных веществах, которые были ранее высказаны Пастером (см. Винная кислота); ниже приводятся факты, которые были найдены непосредственно под влиянием теории и которые ее прямо подтверждают.Как указано, для веществ общей формулы Cabcd, где a, b, c и d представляют нетождественные одноатомные остатки [Таковыми могут быть отдельные атомы или же остатки формул (Cab)l и (Ca2b)r; см. Стереохимия]), возможны два изомера и один полимер; первые являются оптическими антиподами, третий представляет продукт их взаимодействия в эквимолекулярных отношениях; иногда это рацемическое тело, иногда это просто смесь оптических изомеров (см. выше); тело мезовинного типа (Corps inactif du type indédoublable — Пастера, см.) с такой формулой — невозможно. Во время появления учения Фант-Гофф-Ле-Беля имелось, однако, указание Пастера на то, что яблочная кислота — НО—ОС—СН(ОН)—CH2—СО—ОН, получаемая из оптически недеятельной аспарагиновой — HO—OC—CH(NH2)—CH2—CO—OH — не только оптически недеятельна, но и отличается кристаллографическими формами солей от оптически недеятельной же яблочной кисл., получаемой из монобромоянтарной, напр.; попытки Пастера разделить кислоту на оптические антиподы были неудачны, и он признал ее за кислоту мезовинного типа. Исследования Фант-Гоффа, Бремера, Аншютца показали полное тождество всех описанных в литературе оптически недеятельных яблочных кислот, и Бремер посредством соли цинхонина разделил недеятельную кислоту Пастера на правую и левую яблочные кислоты, что и требовалось теорией. — Затем стереохимическая теория, признавая вещества общих формул — Ca3b, Ca2bc симметрично построенными [Об инозите — см. ниже], требует, чтобы они не обладали оптической деятельностью. При возникновении теории этому противоречила оптическая деятельность: пропилового спирта — СН3—СН2—СН2(ОН) (Шансель), стирола (из стиракса) — С6Н5—СН=СН2 (Бертело), β-пиколина — NC5H4—СН3 (Гезекиель) и йодистого триметилэтилстибина — (СН3)3(С2Н5)SbJ (Фридлендер). Оптическая деятельность пропилового спирта оказалась следствием примеси амилового (СН3)(С2Н5)С(Н)(СН2—ОН) [Способ писания предложен Ландольтом для выделения атома углерода, соединенного с 4 различными одноатомными остатками] (Геннйнгер), стирола — примеси каких-то ближе не характеризованных тел (Фант-Гофф, Кракау, фон Миллер, Вегер), β-пиколина — ошибки наблюдения (Ландольт) и йодистого триметилэтилстибина следствием нечистоты употребленного этилового спирта, содержавшего тоже, по-видимому, амиловый спирть (Ле-Бель). Точно так же Вальдену (1898) удалось показать, что оптическая деятельность таннина [Предложенная для него Шиффом формула не содержала асимметричного углерода], найденная Флавицким (1890) и Гюнтером (1895), является следствием химической неоднородности тела, известного под этим названием; наконец, Аберсон нашел, что вращательная способность оксипировиноградной кислоты — СН2(ОН)—СО—СО—ОН объясняется присутствием оптически деятельных азотистых веществ, чистая же кислота не вращает плоскости поляризации. В настоящее время не существует ни одного ациклического соединения, оптическая деятельность которого находилась бы в противоречии с его формулой. Некоторые из таких веществ обладают, однако, слишком слабой оптической деятельностью, которая только с трудом может быть установлена. В случае гидроксилированных соединений оптическая деятельность может быть повышена прибавкою к их растворам солей бериллия (Розенгейм и Итциг), борной кислоты (Био, Пастер), окиси сурьмы, мышьяковистого ангидрида (Пастер, Жернез), окислов молибдена, вольфрама (Жернез) и особенно азотнокислого уранила — UO2(NO3)2+6H2O (Вальден, Дитрих) в присутствии едкой щелочи [Причиной повышения оптической деятельности является образование особых сложных эфиров]. С увеличением числа асимметричных углеродных атомов (см. Стереохимия) в молекуле — число возможных оптических изомеров быстро возрастает и уже начиная с двух асимметричных атомов С для предельного соединения, кроме право- и левовращающих изомеров и их рацемического полимера, возможны и тела мезовинного типа. Первые формулы для вычисления числа изомеров даны Фант-Гоффом; выведены они на основании рассмотрения возможных сочетаний (см.) в предположении свободного вращения атомов (групп), соединенных с углеродом, вокруг атомов последнего (см. Стереохимия). Формулы эти следующие. Пусть n — число асимметричных атомов углерода в молекуле предельного соединения, N — общее число стереоизомеров, Ni — число стереоизомеров оптически недеятельных мезовинного типа и Na — число оптически деятельных изомеров, пары которых являются оптическими антиподами, способными при взаимодействии дать 1/2Na — рацемических форм, тогда в общем случае, при n четном или нечетном и при структурной формуле, не состоящей из тождественных половин, — N = 2n, Na = 2n и Ni = 0, что приводит к следующей таблице:----------------------------------------------------------------------------| n = 1 | 2 | 3 | 4 | 5 | 6 | 7 ||---------------------------------------------------------------------------|| N = Na = 2 | 4 | 8 | 16 | 32 | 64 | 128 |----------------------------------------------------------------------------и т. д. Когда n — четное число и структурная формула состоит из двух половин, то----------------------------------------------------------------| n = | 2 | 4 | 6 | 8 ||---------------------------------------------------------------|| N = | 3 | 10 | 36 | 136 ||---------------------------------------------------------------|| Na = | 2 | 8 | 32 | 128 ||---------------------------------------------------------------|| Ni = | 1 | 2 | 4 | 8 |----------------------------------------------------------------и т. д. Для примера выведем число изомеров при n = 4; если обозначить асимметричные группы буквами A, В, С, D и правую асимметрию знаком "+", а левую знаком "—" [Такое обозначение введено Вант-Гоффом. Для наглядности можно представить себе, что сложное тело образовано взаимодействием право- (+)А и лево- (—)B -вращающих кислот и (+)С и (—)D вращающих оснований. В наиболее простом случае, очевидно, возможны тела: +А+С, —B—D, +A—D и —В+С], то в общем случае 16 изомеров можно представить таким образом:
Фиг. 5. Фиг. 6Верхняя кривая есть кривая температур застывания; нижняя — дает состав кристаллов, выпадающих в самом начале застывания из жидкого сплава; соответственные точки верхней и нижней кривых лежат на горизонтальной линии (ab — фиг. 5 и 6); отрезки ординат (ас — фиг. 5 и 6) между кривыми дают разность температур начала и конца застывания или начала и конца плавления. Благодаря симметрии как максимум, так и минимум приходятся на смеси: 50% d-изомера + 50% l-изомера; в этих точках верхняя и нижняя кривые на самом деле соприкасаются, а это значит, что инактивные кристаллические смеси плавятся при постоянной температуре. Таким образом, псевдорацемия характеризуется непрерывностью кривых плавления и застывания. К оптически деятельным веществам, способным к псевдорацемии типа фиг. 6, Б. Роозебоом относит исследованные Кипингом хлор- и бромангидриды сульфокамфорной кислоты и трибромкарбон из гидрокарбона. Чтобы закончить о рацемических соединениях, необходимо указать, что Поп, Пичей, Гарвей и Рич рекомендуют для разделения рацемических оснований d-α-хлоро- и бромокамфорсульфоновую кислоту — С10Н14ClO·SO3Н и C10H14BrO·SO3H; при помощи этих реактивов им удалось разделить: тетрагидропапаверин — С20Н25NO4, тетрагидрохиналдин — C10H13N, тетрагидропаратолухиналдин — C11H15N, тетрагидро-бета-нафтиламин С10Н12NH2 и камфороксим — С10Н15NOH. Затем Э. Фишер указал, что при активировании слабых амидокислот удобнее всего исходить из бензоилированных производных, которые более постоянны и легче кристаллизуются, чем амидокислоты, и из которых бензоильная группа может быть легко элиминирована. Этим путем с помощью бруцина и стрихнина ему удалось разделить — аланин, аспарагиновую и глутаминовую кислоты и тирозин. Наконец, Марквальд (с Маккензи) и Вальден показали возможность разделения рацемических соединений при взаимодействии их с оптически деятельными веществами на основании различной скорости реакции для d- и l-изомеров, которая возможна потому, что получаемые производные не являются оптически энантиоморфными. Первым удалось получить небольшое количество l-миндальной кислоты (см.) при неполной этерификации l-ментолом r-миндальной кислоты; Вальдену удалось констатировать незначительное разделение при этерификации l-амиловым спиртом бромангидрида r-бромопропионовой кислоты [Вполне подобные опыты Фрейндлера и Симона дали ранее отрицательные результаты.]. Как замечает Э. Фишер, способ по существу тождествен с так называемым биологическим методом разделения оптических антиподов Пастера, так как в последнем случае процесс сводится на взаимодействие рацемического вещества с оптически деятельной энзимой, вырабатываемой данным микроорганизмом.Все приведенные факты находятся в полной независимости от стереохимической гипотезы (см. Стереохимия) Фант-Гоффа и Ле-Беля; они не противоречат ей, но могли быть установлены даже на почве тех представлений об оптически деятельных веществах, которые были ранее высказаны Пастером (см. Винная кислота); ниже приводятся факты, которые были найдены непосредственно под влиянием теории и которые ее прямо подтверждают.Как указано, для веществ общей формулы Cabcd, где a, b, c и d представляют нетождественные одноатомные остатки [Таковыми могут быть отдельные атомы или же остатки формул (Cab)l и (Ca2b)r; см. Стереохимия]), возможны два изомера и один полимер; первые являются оптическими антиподами, третий представляет продукт их взаимодействия в эквимолекулярных отношениях; иногда это рацемическое тело, иногда это просто смесь оптических изомеров (см. выше); тело мезовинного типа (Corps inactif du type indédoublable — Пастера, см.) с такой формулой — невозможно. Во время появления учения Фант-Гофф-Ле-Беля имелось, однако, указание Пастера на то, что яблочная кислота — НО—ОС—СН(ОН)—CH2—СО—ОН, получаемая из оптически недеятельной аспарагиновой — HO—OC—CH(NH2)—CH2—CO—OH — не только оптически недеятельна, но и отличается кристаллографическими формами солей от оптически недеятельной же яблочной кисл., получаемой из монобромоянтарной, напр.; попытки Пастера разделить кислоту на оптические антиподы были неудачны, и он признал ее за кислоту мезовинного типа. Исследования Фант-Гоффа, Бремера, Аншютца показали полное тождество всех описанных в литературе оптически недеятельных яблочных кислот, и Бремер посредством соли цинхонина разделил недеятельную кислоту Пастера на правую и левую яблочные кислоты, что и требовалось теорией. — Затем стереохимическая теория, признавая вещества общих формул — Ca3b, Ca2bc симметрично построенными [Об инозите — см. ниже], требует, чтобы они не обладали оптической деятельностью. При возникновении теории этому противоречила оптическая деятельность: пропилового спирта — СН3—СН2—СН2(ОН) (Шансель), стирола (из стиракса) — С6Н5—СН=СН2 (Бертело), β-пиколина — NC5H4—СН3 (Гезекиель) и йодистого триметилэтилстибина — (СН3)3(С2Н5)SbJ (Фридлендер). Оптическая деятельность пропилового спирта оказалась следствием примеси амилового (СН3)(С2Н5)С(Н)(СН2—ОН) [Способ писания предложен Ландольтом для выделения атома углерода, соединенного с 4 различными одноатомными остатками] (Геннйнгер), стирола — примеси каких-то ближе не характеризованных тел (Фант-Гофф, Кракау, фон Миллер, Вегер), β-пиколина — ошибки наблюдения (Ландольт) и йодистого триметилэтилстибина следствием нечистоты употребленного этилового спирта, содержавшего тоже, по-видимому, амиловый спирть (Ле-Бель). Точно так же Вальдену (1898) удалось показать, что оптическая деятельность таннина [Предложенная для него Шиффом формула не содержала асимметричного углерода], найденная Флавицким (1890) и Гюнтером (1895), является следствием химической неоднородности тела, известного под этим названием; наконец, Аберсон нашел, что вращательная способность оксипировиноградной кислоты — СН2(ОН)—СО—СО—ОН объясняется присутствием оптически деятельных азотистых веществ, чистая же кислота не вращает плоскости поляризации. В настоящее время не существует ни одного ациклического соединения, оптическая деятельность которого находилась бы в противоречии с его формулой. Некоторые из таких веществ обладают, однако, слишком слабой оптической деятельностью, которая только с трудом может быть установлена. В случае гидроксилированных соединений оптическая деятельность может быть повышена прибавкою к их растворам солей бериллия (Розенгейм и Итциг), борной кислоты (Био, Пастер), окиси сурьмы, мышьяковистого ангидрида (Пастер, Жернез), окислов молибдена, вольфрама (Жернез) и особенно азотнокислого уранила — UO2(NO3)2+6H2O (Вальден, Дитрих) в присутствии едкой щелочи [Причиной повышения оптической деятельности является образование особых сложных эфиров]. С увеличением числа асимметричных углеродных атомов (см. Стереохимия) в молекуле — число возможных оптических изомеров быстро возрастает и уже начиная с двух асимметричных атомов С для предельного соединения, кроме право- и левовращающих изомеров и их рацемического полимера, возможны и тела мезовинного типа. Первые формулы для вычисления числа изомеров даны Фант-Гоффом; выведены они на основании рассмотрения возможных сочетаний (см.) в предположении свободного вращения атомов (групп), соединенных с углеродом, вокруг атомов последнего (см. Стереохимия). Формулы эти следующие. Пусть n — число асимметричных атомов углерода в молекуле предельного соединения, N — общее число стереоизомеров, Ni — число стереоизомеров оптически недеятельных мезовинного типа и Na — число оптически деятельных изомеров, пары которых являются оптическими антиподами, способными при взаимодействии дать 1/2Na — рацемических форм, тогда в общем случае, при n четном или нечетном и при структурной формуле, не состоящей из тождественных половин, — N = 2n, Na = 2n и Ni = 0, что приводит к следующей таблице:----------------------------------------------------------------------------| n = 1 | 2 | 3 | 4 | 5 | 6 | 7 ||---------------------------------------------------------------------------|| N = Na = 2 | 4 | 8 | 16 | 32 | 64 | 128 |----------------------------------------------------------------------------и т. д. Когда n — четное число и структурная формула состоит из двух половин, то----------------------------------------------------------------| n = | 2 | 4 | 6 | 8 ||---------------------------------------------------------------|| N = | 3 | 10 | 36 | 136 ||---------------------------------------------------------------|| Na = | 2 | 8 | 32 | 128 ||---------------------------------------------------------------|| Ni = | 1 | 2 | 4 | 8 |----------------------------------------------------------------и т. д. Для примера выведем число изомеров при n = 4; если обозначить асимметричные группы буквами A, В, С, D и правую асимметрию знаком "+", а левую знаком "—" [Такое обозначение введено Вант-Гоффом. Для наглядности можно представить себе, что сложное тело образовано взаимодействием право- (+)А и лево- (—)B -вращающих кислот и (+)С и (—)D вращающих оснований. В наиболее простом случае, очевидно, возможны тела: +А+С, —B—D, +A—D и —В+С], то в общем случае 16 изомеров можно представить таким образом: Когда же формула делима пополам, то A = D, В = С; случаи 2, 3, 4, 6, 8 и 12 тождественны тогда со случаями 9, 5, 13, 11, 15 и 14, и число остающихся изомеров равно 10, а именно:
Когда же формула делима пополам, то A = D, В = С; случаи 2, 3, 4, 6, 8 и 12 тождественны тогда со случаями 9, 5, 13, 11, 15 и 14, и число остающихся изомеров равно 10, а именно: [Конфигурации (см. Стереохимия) 1—4, 2—3, 5—9, 6—10 являются оптическими антиподами, 7 и 8 тела мезовинного типа]. Наконец, когда n нечетное число, а структурная формула хотя и не делится пополам, но симметрична [Ландольт вполне правильно замечает, что средний углеродный атом таких формул должен считаться псевдоасимметричным] по отношению к среднему атому углерода, тоN = 2n—1 , Na = 2n—1 — 2[(n—1)/2] и Ni = 2[(n—1)/2],что дает--------------------------------------------------| n = | 3 | 5 | 7 | 9 ||-------------------------------------------------|| N = | 4 | 16 | 64 | 256 ||-------------------------------------------------|| Na = | 2 | 12 | 56 | 240 ||-------------------------------------------------|| Ni = | 2 | 4 | 8 | 16 |--------------------------------------------------Очевидно, что только в простейших случаях можно изучить все изомеры, ожидаемые теорией [К числу N надо прибавить еще (Na)/2 число рацемических форм]. Ввиду того, что при сложных формулах сокращенное обозначение асимметричных остатков знаками + и -является сбивчивым, Э. Фишер предложил в таких случаях пользоваться проекциями стереометрических формул на бумаге. Для этого сначала при помощи каучуковых моделей Фридлендера (см. Стереохимия) строят желаемую формулу, затем кладут ее на бумагу так, чтобы все атомы углерода приходились на одной линии, а соединенные с ними атомы и остатки (очень часто это водородные атомы и водные остатки) находились бы над плоскостью бумаги; прямоугольная проекция их и дает искомую конфигурацию. Цепь углеродных атомов можно обозначить прямой линией (В. Мейер и Якобсон), а водородные атомы и водные остатки обозначить — первые кружками, а вторые — крестиками (Ландольт), и таким образом получаются следующие стереохимические формулы для d- и l-глюкоз:
[Конфигурации (см. Стереохимия) 1—4, 2—3, 5—9, 6—10 являются оптическими антиподами, 7 и 8 тела мезовинного типа]. Наконец, когда n нечетное число, а структурная формула хотя и не делится пополам, но симметрична [Ландольт вполне правильно замечает, что средний углеродный атом таких формул должен считаться псевдоасимметричным] по отношению к среднему атому углерода, тоN = 2n—1 , Na = 2n—1 — 2[(n—1)/2] и Ni = 2[(n—1)/2],что дает--------------------------------------------------| n = | 3 | 5 | 7 | 9 ||-------------------------------------------------|| N = | 4 | 16 | 64 | 256 ||-------------------------------------------------|| Na = | 2 | 12 | 56 | 240 ||-------------------------------------------------|| Ni = | 2 | 4 | 8 | 16 |--------------------------------------------------Очевидно, что только в простейших случаях можно изучить все изомеры, ожидаемые теорией [К числу N надо прибавить еще (Na)/2 число рацемических форм]. Ввиду того, что при сложных формулах сокращенное обозначение асимметричных остатков знаками + и -является сбивчивым, Э. Фишер предложил в таких случаях пользоваться проекциями стереометрических формул на бумаге. Для этого сначала при помощи каучуковых моделей Фридлендера (см. Стереохимия) строят желаемую формулу, затем кладут ее на бумагу так, чтобы все атомы углерода приходились на одной линии, а соединенные с ними атомы и остатки (очень часто это водородные атомы и водные остатки) находились бы над плоскостью бумаги; прямоугольная проекция их и дает искомую конфигурацию. Цепь углеродных атомов можно обозначить прямой линией (В. Мейер и Якобсон), а водородные атомы и водные остатки обозначить — первые кружками, а вторые — крестиками (Ландольт), и таким образом получаются следующие стереохимические формулы для d- и l-глюкоз: Какую формулу приписать d- и какую l-глюкозе, зависит от произвола (Фишер принимает верхнюю формулу за выражение стереохимического строения d-, а нижнюю — l-глюкозы), но что глюкозам принадлежат именно эти формулы, а не какие-нибудь другие из 16 возможных для веществ с 4-мя асимметричными атомами углерода (см. выше табл. I), это установлено Э. Фишером с довольно значительной степенью вероятности. Ход его рассуждения был следующий. 1) Для виноградного сахара и его изомеров теория предвидит 16 стереоизомерных форм, а для их производных, у которых в силу структурной симметрии формула делима пополам, только 10 изомеров (см. данные выше табл.). 2) Сахарная кислота — НО—ОС—СН(ОН)—СН(ОН)—СН(ОН)—CH(ОН)—СО—ОН получается как из виноградного сахара, так и из d-гулозы (Фишер и Пилоти; см. Глюкозы), а из этого следует, что формула ее должна отвечать одной из формул, обозначенных в таблице II цифрами 5—10, потому что только они допускают возможность образования одного и того же вещества из двух стереоизомеров [Это видно как из таблицы II, так и из сравнения ее с таблицей I: оно показывает, что изомеры 2—9, 3—5 и т. д. сливаются при тождестве А с D, B с С]. 3) Между этими формулами — формулы 7 и 8 должны быть исключены, как отвечающие телам мезовинного типа (см.), виноградный же сахар, гулоза и сахарные кислоты оптически деятельны. 4) Ближайшее рассмотрение показывает, что глюкоза не может иметь ни 6, ни 10 формулы (табл.); все известные факты говорят за полное структурное тождество ее с маннозой, а существование для них одного озазона, способность арабинозы НО—СН2—СН(ОН)—СН(ОН)—СН(ОН)—СОН давать при действии синильной кислоты одновременно l-манноновую и l-глюконовую кислоты, способность этих кислот переходить друг в друга при нагревании с хинолином — все эти реакции говорят за то, что они отличаются друг от друга только пространственным расположением атома водорода и водного остатка при том углеродном атоме, который в формуле НО—СН2—[СН(ОН)]3—СН*(ОН)—СОН [Знаком * отмечен в этой и следующих формулах тот углеродный атом, при котором в глюкозе и маннозе разным образом распределены стереохимически H и ОН.] стоит рядом с альдегидной группой и обозначен звездочкой, а потому если для сахарной кислоты (и для глюкозы) принять последовательность асимметрии, выражаемую формулами 6 или 10, т. е. +*—++ (№ 6) или -+——* (№ 10), то манносахарной кислоте (и маннозе) должно приписать формулы: —*—++ (№ 7) или —+—+* (№ 8), а это формулы тел мезовинного типа, что не отвечает действительности, так как маннит и манносахарная кислота оптически деятельны (известны d-, l- и r- формы). Таким образом, для d- и l-сахарных кислот остаются только формулы 5 и 9, т. е. —+++ и +———. 5) d-сахарной кислоте отвечают 2 альдозы: глюкоза и гулоза, а след., их формулы могут быть:
Какую формулу приписать d- и какую l-глюкозе, зависит от произвола (Фишер принимает верхнюю формулу за выражение стереохимического строения d-, а нижнюю — l-глюкозы), но что глюкозам принадлежат именно эти формулы, а не какие-нибудь другие из 16 возможных для веществ с 4-мя асимметричными атомами углерода (см. выше табл. I), это установлено Э. Фишером с довольно значительной степенью вероятности. Ход его рассуждения был следующий. 1) Для виноградного сахара и его изомеров теория предвидит 16 стереоизомерных форм, а для их производных, у которых в силу структурной симметрии формула делима пополам, только 10 изомеров (см. данные выше табл.). 2) Сахарная кислота — НО—ОС—СН(ОН)—СН(ОН)—СН(ОН)—CH(ОН)—СО—ОН получается как из виноградного сахара, так и из d-гулозы (Фишер и Пилоти; см. Глюкозы), а из этого следует, что формула ее должна отвечать одной из формул, обозначенных в таблице II цифрами 5—10, потому что только они допускают возможность образования одного и того же вещества из двух стереоизомеров [Это видно как из таблицы II, так и из сравнения ее с таблицей I: оно показывает, что изомеры 2—9, 3—5 и т. д. сливаются при тождестве А с D, B с С]. 3) Между этими формулами — формулы 7 и 8 должны быть исключены, как отвечающие телам мезовинного типа (см.), виноградный же сахар, гулоза и сахарные кислоты оптически деятельны. 4) Ближайшее рассмотрение показывает, что глюкоза не может иметь ни 6, ни 10 формулы (табл.); все известные факты говорят за полное структурное тождество ее с маннозой, а существование для них одного озазона, способность арабинозы НО—СН2—СН(ОН)—СН(ОН)—СН(ОН)—СОН давать при действии синильной кислоты одновременно l-манноновую и l-глюконовую кислоты, способность этих кислот переходить друг в друга при нагревании с хинолином — все эти реакции говорят за то, что они отличаются друг от друга только пространственным расположением атома водорода и водного остатка при том углеродном атоме, который в формуле НО—СН2—[СН(ОН)]3—СН*(ОН)—СОН [Знаком * отмечен в этой и следующих формулах тот углеродный атом, при котором в глюкозе и маннозе разным образом распределены стереохимически H и ОН.] стоит рядом с альдегидной группой и обозначен звездочкой, а потому если для сахарной кислоты (и для глюкозы) принять последовательность асимметрии, выражаемую формулами 6 или 10, т. е. +*—++ (№ 6) или -+——* (№ 10), то манносахарной кислоте (и маннозе) должно приписать формулы: —*—++ (№ 7) или —+—+* (№ 8), а это формулы тел мезовинного типа, что не отвечает действительности, так как маннит и манносахарная кислота оптически деятельны (известны d-, l- и r- формы). Таким образом, для d- и l-сахарных кислот остаются только формулы 5 и 9, т. е. —+++ и +———. 5) d-сахарной кислоте отвечают 2 альдозы: глюкоза и гулоза, а след., их формулы могут быть:![но для того, чтобы решить, какая из этих формул принадлежит глюкозе, а какая гулозе, необходимо обратиться к рассмотрению стереохимического строения арабинозы и ксилозы, из которых могут быть получены методом цианистых соединений — глюкоза и гулоза. Альдегидная группа, их характеризующая, является результатом присоединения элементов оксиметилена к альдозам: арабинозе и ксилозе [На самом деле присоединяется сначала синильная кислота; полученный нитрил гидратацией переводится в кислоту, а последняя через лактон восстановляется в альдегид, следовательно, группа (CN) синильной кислоты — HCN — переходит через —СО(ОН) в (СОН)], а, следовательно, строение арабинозы и ксилозы может выражаться формулами:](/pictures/brokgauz_efron/b62_614-1.jpg) но для того, чтобы решить, какая из этих формул принадлежит глюкозе, а какая гулозе, необходимо обратиться к рассмотрению стереохимического строения арабинозы и ксилозы, из которых могут быть получены методом цианистых соединений — глюкоза и гулоза. Альдегидная группа, их характеризующая, является результатом присоединения элементов оксиметилена к альдозам: арабинозе и ксилозе [На самом деле присоединяется сначала синильная кислота; полученный нитрил гидратацией переводится в кислоту, а последняя через лактон восстановляется в альдегид, следовательно, группа (CN) синильной кислоты — HCN — переходит через —СО(ОН) в (СОН)], а, следовательно, строение арабинозы и ксилозы может выражаться формулами:
но для того, чтобы решить, какая из этих формул принадлежит глюкозе, а какая гулозе, необходимо обратиться к рассмотрению стереохимического строения арабинозы и ксилозы, из которых могут быть получены методом цианистых соединений — глюкоза и гулоза. Альдегидная группа, их характеризующая, является результатом присоединения элементов оксиметилена к альдозам: арабинозе и ксилозе [На самом деле присоединяется сначала синильная кислота; полученный нитрил гидратацией переводится в кислоту, а последняя через лактон восстановляется в альдегид, следовательно, группа (CN) синильной кислоты — HCN — переходит через —СО(ОН) в (СОН)], а, следовательно, строение арабинозы и ксилозы может выражаться формулами: [Синтезированы были первыми не d-глюкоза и гулоза, а их l-изомеры, и потому следовало бы взять формулы энантиоморфных арабинозы и ксилозы; аргументация этим не была бы, однако, изменена, так как Воль получил d-арабинозу из d-глюкозы.]. Вопрос о том, какая формула принадлежит какой пентозе — решается окислением их в триоксиглутаровые кислоты:
[Синтезированы были первыми не d-глюкоза и гулоза, а их l-изомеры, и потому следовало бы взять формулы энантиоморфных арабинозы и ксилозы; аргументация этим не была бы, однако, изменена, так как Воль получил d-арабинозу из d-глюкозы.]. Вопрос о том, какая формула принадлежит какой пентозе — решается окислением их в триоксиглутаровые кислоты: дело в том, что благодаря псевдоасимметрии среднего атома углерода в этих формулах, первая из приведенных конфигураций не может дать энантиоморфной формы [Наглядно вопрос решается только при посредстве тетраэдрических моделей] и потому должна отвечать веществу оптически недеятельному — мезовинного типа, а вторая мыслима в двух энантиоморфных формах и потому должна отвчать оптически деятельной триоксиглутаровой кислоте, существующей в виде d- и l- изомеров и r-полимера. Опыт оправдывает это предвидение теории, так как триоксиглутаровая кислота, получаемая из d-арабинозы, оптически деятельна (О. Руфф) и существует в виде d- и l-изомеров (Руфф, Килиани) и r-полимера (Руфф), а триоксиглутаровая кислота, получаемая из ксилозы, оптически недеятельна и не могла быть разложена на оптические изомеры (Э. Фишер, Э. Фишер и О. Пилотти, О. Руфф), а потому должна считаться принадлежащей к мезовинному типу. Таким образом выводится, что формула
дело в том, что благодаря псевдоасимметрии среднего атома углерода в этих формулах, первая из приведенных конфигураций не может дать энантиоморфной формы [Наглядно вопрос решается только при посредстве тетраэдрических моделей] и потому должна отвечать веществу оптически недеятельному — мезовинного типа, а вторая мыслима в двух энантиоморфных формах и потому должна отвчать оптически деятельной триоксиглутаровой кислоте, существующей в виде d- и l- изомеров и r-полимера. Опыт оправдывает это предвидение теории, так как триоксиглутаровая кислота, получаемая из d-арабинозы, оптически деятельна (О. Руфф) и существует в виде d- и l-изомеров (Руфф, Килиани) и r-полимера (Руфф), а триоксиглутаровая кислота, получаемая из ксилозы, оптически недеятельна и не могла быть разложена на оптические изомеры (Э. Фишер, Э. Фишер и О. Пилотти, О. Руфф), а потому должна считаться принадлежащей к мезовинному типу. Таким образом выводится, что формула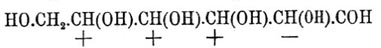 принадлежит d-глюкозе, а формула
принадлежит d-глюкозе, а формула — d-гулозе; их оптическим антиподам должны принадлежать формулы:
— d-гулозе; их оптическим антиподам должны принадлежать формулы: Какое выбрать пространственное расположение для выражения стереохимического строения правовращающих веществ, зависит от нашего произвола: но раз мы такое расположение выберем, то зеркальное изображение, с ним несовместимое, должно считаться отвечающим левовращающему веществу. Э. Фишер, впрочем, держится того мнения, и оно повторяется большинством современных учебников по органической химии, что ему удалось это пространственное расположение найти (1896 г.) [Вант-Гофф (Zweites Heft, "Vorlesungen ü. theoretieche u. physikalische Chemie,1899) умалчивает о доказательстве Э. Фишера и напротив, подчеркивает невозможность такой задачи]. В основании его соображений лежат следующие факты. d-рамноза (изодульцит, метилпентоза, см. Гидраты углерода и Глюкозы) дает при окислении азотной кислотою ту же l-триоксиглутаровую кислоту (Виль и Петерс), которая получается и из l-арабинозы (Килиани); реакция заключается в отщеплении метильной группы и окислении рядом стоящего с нею атома углерода в карбоксильную группу; другая карбоксильная группа образуется на счет альдегидной группы — (СОН)' — рамнозы. С другой стороны, рамноза (методом Воля) [Из альдозы действием гидроксиламина получают оксим: R—CH(ОН)—СОН + NH2O = R—CH(ОН)—СН=N(OH) + H2O; оксим при действии водоотнимающих веществ превращают в нитрил: R—CH(OH)—CH=N(OH) — Н2O = R—CH(OH)—CN, который (в присутствии окиси серебра), теряя элементы синильной кислоты, дает новую альдозу: R—CH(OH)CN — HCN = R—COH, отличающуюся от исходной на элементы оксиметилена.] превращена Э. Фишером и Пинкусом в метилтетрозу, а эта последняя, при окислении азотной кислотою, в d-винную кислоту, находящуюся в том же отношении к метилтетрозе, в котором триоксиглутаровая находится к d-рамнозе. Если l-триоксиглутаровой кислоте приписать формулу
Какое выбрать пространственное расположение для выражения стереохимического строения правовращающих веществ, зависит от нашего произвола: но раз мы такое расположение выберем, то зеркальное изображение, с ним несовместимое, должно считаться отвечающим левовращающему веществу. Э. Фишер, впрочем, держится того мнения, и оно повторяется большинством современных учебников по органической химии, что ему удалось это пространственное расположение найти (1896 г.) [Вант-Гофф (Zweites Heft, "Vorlesungen ü. theoretieche u. physikalische Chemie,1899) умалчивает о доказательстве Э. Фишера и напротив, подчеркивает невозможность такой задачи]. В основании его соображений лежат следующие факты. d-рамноза (изодульцит, метилпентоза, см. Гидраты углерода и Глюкозы) дает при окислении азотной кислотою ту же l-триоксиглутаровую кислоту (Виль и Петерс), которая получается и из l-арабинозы (Килиани); реакция заключается в отщеплении метильной группы и окислении рядом стоящего с нею атома углерода в карбоксильную группу; другая карбоксильная группа образуется на счет альдегидной группы — (СОН)' — рамнозы. С другой стороны, рамноза (методом Воля) [Из альдозы действием гидроксиламина получают оксим: R—CH(ОН)—СОН + NH2O = R—CH(ОН)—СН=N(OH) + H2O; оксим при действии водоотнимающих веществ превращают в нитрил: R—CH(OH)—CH=N(OH) — Н2O = R—CH(OH)—CN, который (в присутствии окиси серебра), теряя элементы синильной кислоты, дает новую альдозу: R—CH(OH)CN — HCN = R—COH, отличающуюся от исходной на элементы оксиметилена.] превращена Э. Фишером и Пинкусом в метилтетрозу, а эта последняя, при окислении азотной кислотою, в d-винную кислоту, находящуюся в том же отношении к метилтетрозе, в котором триоксиглутаровая находится к d-рамнозе. Если l-триоксиглутаровой кислоте приписать формулу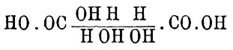 (Э. Фишер и Морелль)[Э. Фишер просмотрел, что эта формула является произвольной], то для строения dрамнозы наиболее вероятна формула:
(Э. Фишер и Морелль)[Э. Фишер просмотрел, что эта формула является произвольной], то для строения dрамнозы наиболее вероятна формула: , а для стереохимического строения метилтетрозы формула:
, а для стереохимического строения метилтетрозы формула:  , причем окисление ее в d-винную можно представить такой схемой:
, причем окисление ее в d-винную можно представить такой схемой: каковой устанавливается и строение d-винной кислоты. А так как, с другой стороны, d-винная кислота (наряду со щавелевой) образуются при окислении виноградного сахара (d-глюкозы), то в связи с только что установленной для последней формулой:
каковой устанавливается и строение d-винной кислоты. А так как, с другой стороны, d-винная кислота (наряду со щавелевой) образуются при окислении виноградного сахара (d-глюкозы), то в связи с только что установленной для последней формулой: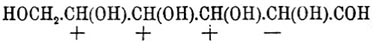 приходится это превращение выразить таким образом:
приходится это превращение выразить таким образом: [Несомненно, что если бы Э. Фишер приписал l-триоксиглутаровой кислоте формулу ее зеркального изображения, то и все остальные формулы соответственным образом изменились бы, вполне отвечая опытным данным] и, след., фиксировать стереохимические формулы: d-винной кислоты, d-глюкозы, их оптических антиподов, d- и l-гулозы и т. д. Необходимо обратить внимание, что при всем вышеизложенном Э. Фишером ни разу не принималась во внимание возможность перегруппировок, а такому допущению противоречат многие данные. Так: Лобри де Брейн и Альберда фан Экенштейн наблюдали, что под влиянием щелочей происходят изомерные превращения мальтозы, лактозы и мелибиозы; Скрауп нашел, что цинхонин изомезируется в α-изоцинхонин в присутствии галоидоводородов (HCl, HBr и HJ); еще важнее наблюдения в этой области П. Вальдена, показавшего возможность кругового процесса, передаваемого такой схемой:
[Несомненно, что если бы Э. Фишер приписал l-триоксиглутаровой кислоте формулу ее зеркального изображения, то и все остальные формулы соответственным образом изменились бы, вполне отвечая опытным данным] и, след., фиксировать стереохимические формулы: d-винной кислоты, d-глюкозы, их оптических антиподов, d- и l-гулозы и т. д. Необходимо обратить внимание, что при всем вышеизложенном Э. Фишером ни разу не принималась во внимание возможность перегруппировок, а такому допущению противоречат многие данные. Так: Лобри де Брейн и Альберда фан Экенштейн наблюдали, что под влиянием щелочей происходят изомерные превращения мальтозы, лактозы и мелибиозы; Скрауп нашел, что цинхонин изомезируется в α-изоцинхонин в присутствии галоидоводородов (HCl, HBr и HJ); еще важнее наблюдения в этой области П. Вальдена, показавшего возможность кругового процесса, передаваемого такой схемой: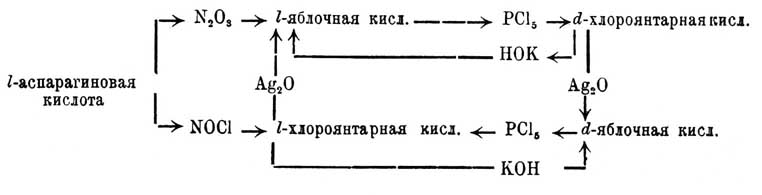 Схема эта наглядно показывает, что различные реактивы далеко не равноценны в стереохимическом отношении; между тем как PCl5 (и PBr5), а также гидрат окиси калия (и гидраты окисей лития, натрия, рубидия, бария, меди, кадмия, свинца, олова и водный раствор аммиака) позволают от l-яблочной кислоты перейти к d-хлороянтарной и обратно любое число раз, — оказывается, что окись и гидрат окиси серебра Ag2O и AgOH (а также окиси и гидраты окисей ртути, таллия и палладия) позволяют переход от d-хлороянтарной к d-яблочной и от l-хлороянтарной к l-яблочной. Вопрос о том, какие реактивы должно считать стереохимически нормальными, окончательно пока не может быть решен. П. Вальден, по-видимому, склоняется к признанию таковыми PCl5 и КОН и веществ, им равноценных [Поводом является ненормальность (?) реакций серебряных солей, сказывающаяся в образовании изонитрилов при реакции RJ + AgCN = R'(CN) + AgJ и нитросоединений при действии азотистокислого серебра на те же йодюры: RJ + AgNO2 = RNO2 + AgJ. Не следует забывать, что азотистокалиевая соль дает с хлороуксусной — нитроуксусную]. Число известных оптически деятельных предельных и непредельных веществ громадно (Ландольт в 1898 г. их насчитывал более 700); в большинстве случаев их взаимные отношения недостаточно твердо установлены, но до сих пор опытные данные всегда подтверждали число предвидимых стереохимией изомеров; так, выше было указано, что при двух асимметричных углеродах возможны четыре стереизомера, и Либерманом получены четыре дибромокоричных кислоты:
Схема эта наглядно показывает, что различные реактивы далеко не равноценны в стереохимическом отношении; между тем как PCl5 (и PBr5), а также гидрат окиси калия (и гидраты окисей лития, натрия, рубидия, бария, меди, кадмия, свинца, олова и водный раствор аммиака) позволают от l-яблочной кислоты перейти к d-хлороянтарной и обратно любое число раз, — оказывается, что окись и гидрат окиси серебра Ag2O и AgOH (а также окиси и гидраты окисей ртути, таллия и палладия) позволяют переход от d-хлороянтарной к d-яблочной и от l-хлороянтарной к l-яблочной. Вопрос о том, какие реактивы должно считать стереохимически нормальными, окончательно пока не может быть решен. П. Вальден, по-видимому, склоняется к признанию таковыми PCl5 и КОН и веществ, им равноценных [Поводом является ненормальность (?) реакций серебряных солей, сказывающаяся в образовании изонитрилов при реакции RJ + AgCN = R'(CN) + AgJ и нитросоединений при действии азотистокислого серебра на те же йодюры: RJ + AgNO2 = RNO2 + AgJ. Не следует забывать, что азотистокалиевая соль дает с хлороуксусной — нитроуксусную]. Число известных оптически деятельных предельных и непредельных веществ громадно (Ландольт в 1898 г. их насчитывал более 700); в большинстве случаев их взаимные отношения недостаточно твердо установлены, но до сих пор опытные данные всегда подтверждали число предвидимых стереохимией изомеров; так, выше было указано, что при двух асимметричных углеродах возможны четыре стереизомера, и Либерманом получены четыре дибромокоричных кислоты:![одна пара получается из коричной, а другая из аллокоричной (см. Рацемия) кислоты и т. д. Об усложнении оптической изомерии в случае присутствия двойной связи см. Рицинолевая и рицинэлаидиновая кислоты и Терпены. — Не поддается пока стереохимическому объяснению различная величина оптической деятельности структурных изомеров, так как попытки Ги и Крум-Броуна отыскать зависимость между величиною и направлением вращения и массою одноатомных групп, связанных с асимметрическим углеродным атомом, оказались вполне неудачными (главная заслуга в выяснении этого вопроса принадлежит П. Вальдену); затем необъясненными надо считать явления мультиротации и изменения величины оптической деятельности и даже перемены ее знака для растворов многих оптически деятельных веществ в зависимости от концентрации этих растворов (ср. Яблочная кислота [Явления во многих отношениях напоминают то, что наблюдается для диамагнитных и аэрамагнитных тел]).](/pictures/brokgauz_efron/b62_616-1.jpg) одна пара получается из коричной, а другая из аллокоричной (см. Рацемия) кислоты и т. д. Об усложнении оптической изомерии в случае присутствия двойной связи см. Рицинолевая и рицинэлаидиновая кислоты и Терпены. — Не поддается пока стереохимическому объяснению различная величина оптической деятельности структурных изомеров, так как попытки Ги и Крум-Броуна отыскать зависимость между величиною и направлением вращения и массою одноатомных групп, связанных с асимметрическим углеродным атомом, оказались вполне неудачными (главная заслуга в выяснении этого вопроса принадлежит П. Вальдену); затем необъясненными надо считать явления мультиротации и изменения величины оптической деятельности и даже перемены ее знака для растворов многих оптически деятельных веществ в зависимости от концентрации этих растворов (ср. Яблочная кислота [Явления во многих отношениях напоминают то, что наблюдается для диамагнитных и аэрамагнитных тел]).Оптическая изомерия азота впервые установлена Ле-Белем, который, культивируя Penicillium в растворе хлористого метилэтилпропилизобутиламмония Cl—N(CH3)(C2H5)(C3H7)(C4H9), выделил левый оптически деятельный изомер и получил для него оптически деятельные: хлороплатинат, хлоромеркурат и уксуснокислую соль. Даже под влиянием слабых кислот вещество легко рацемизируется. Ле-Белем же получен левый хлористый этилпропилизобутилизоамиламмоний. Дальнейшим подтверждением возможности оптически деятельных соединений азота являются наблюдения Попа и Пичея; подвергая действию серебряной соли d-камфорсульфоновой кисл. (ср. выше) йодистый α-бензилфенилаллилметиламмоний [Это вещество получено Ведекиндом в двух видоизменениях: α-, плав. при 140—149° и β-, плав. при 158—159°]: СОС9Н15SO3Ag + JN(C6H5CH2)(С6H5)(С3H5)(CH3) = AgJ + N(C7H7)(С6Н5)(C8H5)(CH3)—C10H15OSO3, они получили частично рацемическое соединение, которое кристаллизацией (из смеси ацетона и уксусного эфира) удалось разделить на правый и левый оптические антиподы; регенерированные йодистые соединения плавятся при 145°—147° и обладают вращением d—[α]D = +52,5°, а 1—[α]D = —51,4°.II. Геометрические изомеры. Принимая, что в метане и его производных атом углерода лежит в центре правильного тетраэдра, по углам которого расположены соединенные с углеродом атомы (одноатомные группы), мы должны представить себе этан в виде двух тетраэдров, соприкасающихся одной из вершин; атомы водорода занимают свободные углы. Если бы такая система (фиг. 7) была неподвижна, то ее можно было бы отождествить с трехгранной призмой; атомы водорода занимали бы в этане то положение, которое им придается призматической формулой бензола Ладенбурга, что должно бы в свою очередь вести к существованию по крайней мере трех изомерных двузамещенных производных формулы C2H4X2, одного структурной формулы: СНХ2—СН3 (фиг. 8) и двух стереоизомерных формулы: СН2Х—СН2Х (фиг. 9 и 10), из которых последний должен представлять рацемическое тело.
одна пара получается из коричной, а другая из аллокоричной (см. Рацемия) кислоты и т. д. Об усложнении оптической изомерии в случае присутствия двойной связи см. Рицинолевая и рицинэлаидиновая кислоты и Терпены. — Не поддается пока стереохимическому объяснению различная величина оптической деятельности структурных изомеров, так как попытки Ги и Крум-Броуна отыскать зависимость между величиною и направлением вращения и массою одноатомных групп, связанных с асимметрическим углеродным атомом, оказались вполне неудачными (главная заслуга в выяснении этого вопроса принадлежит П. Вальдену); затем необъясненными надо считать явления мультиротации и изменения величины оптической деятельности и даже перемены ее знака для растворов многих оптически деятельных веществ в зависимости от концентрации этих растворов (ср. Яблочная кислота [Явления во многих отношениях напоминают то, что наблюдается для диамагнитных и аэрамагнитных тел]).Оптическая изомерия азота впервые установлена Ле-Белем, который, культивируя Penicillium в растворе хлористого метилэтилпропилизобутиламмония Cl—N(CH3)(C2H5)(C3H7)(C4H9), выделил левый оптически деятельный изомер и получил для него оптически деятельные: хлороплатинат, хлоромеркурат и уксуснокислую соль. Даже под влиянием слабых кислот вещество легко рацемизируется. Ле-Белем же получен левый хлористый этилпропилизобутилизоамиламмоний. Дальнейшим подтверждением возможности оптически деятельных соединений азота являются наблюдения Попа и Пичея; подвергая действию серебряной соли d-камфорсульфоновой кисл. (ср. выше) йодистый α-бензилфенилаллилметиламмоний [Это вещество получено Ведекиндом в двух видоизменениях: α-, плав. при 140—149° и β-, плав. при 158—159°]: СОС9Н15SO3Ag + JN(C6H5CH2)(С6H5)(С3H5)(CH3) = AgJ + N(C7H7)(С6Н5)(C8H5)(CH3)—C10H15OSO3, они получили частично рацемическое соединение, которое кристаллизацией (из смеси ацетона и уксусного эфира) удалось разделить на правый и левый оптические антиподы; регенерированные йодистые соединения плавятся при 145°—147° и обладают вращением d—[α]D = +52,5°, а 1—[α]D = —51,4°.II. Геометрические изомеры. Принимая, что в метане и его производных атом углерода лежит в центре правильного тетраэдра, по углам которого расположены соединенные с углеродом атомы (одноатомные группы), мы должны представить себе этан в виде двух тетраэдров, соприкасающихся одной из вершин; атомы водорода занимают свободные углы. Если бы такая система (фиг. 7) была неподвижна, то ее можно было бы отождествить с трехгранной призмой; атомы водорода занимали бы в этане то положение, которое им придается призматической формулой бензола Ладенбурга, что должно бы в свою очередь вести к существованию по крайней мере трех изомерных двузамещенных производных формулы C2H4X2, одного структурной формулы: СНХ2—СН3 (фиг. 8) и двух стереоизомерных формулы: СН2Х—СН2Х (фиг. 9 и 10), из которых последний должен представлять рацемическое тело.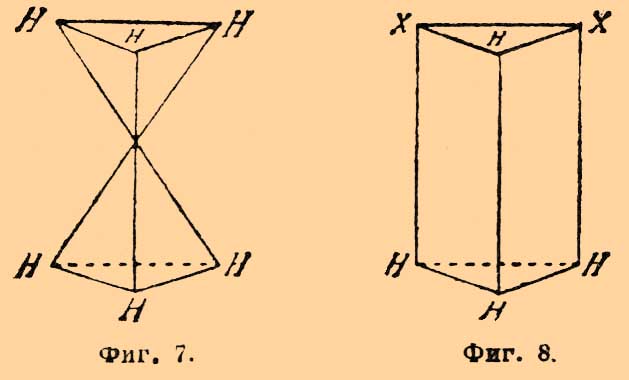 Фиг. 7. Фиг. 8
Фиг. 7. Фиг. 8 Фиг. 9. Фиг. 10Так как СН2Х—СН2Х существует только в одном видоизменении [Единственным исключением, правда, до сих пор не окончательно установленным, является открытие Аберсоном в растениях сем. Crassulaceae яблочной кислоты НО—СО—ОН(ОН)—СН2—СО—ОН, отличной от обыкновенной, ее антипода и их рацемического полимера. Для тела с одним асимметричным атомом углерода другие изомеры при свободе вращения углеродных систем немыслимы], то должно допустить, что тетраэдры, находящиеся в положении фиг. 7, обладают свободным вращением вокруг оси, их соединяющей; все формулы изомерных глюкоз и их производных, рассмотренные ранее, выведены именно в предположении такого свободного вращения. Совершенно противоположное отношение приходится принять для двух атомов углерода, соединенных двойною связью, т. е. для производных этилена формулы abC:Ccd, где а, b, с и d представляют любые одноатомные остатки (атомы). С точки зрения тетраэдрической гипотезы (Вант-Гоффа) соединение двух атомов углерода двойною связью приходится изображать двумя тетраэдрами, соединенными уже не вершинами, а ребром (фиг. 11), и Вант-Гофф с самого начала принял, что в таких системах относительное положение групп, соединенных с углеродом, фиксировано и что они расположены в одной плоскости.
Фиг. 9. Фиг. 10Так как СН2Х—СН2Х существует только в одном видоизменении [Единственным исключением, правда, до сих пор не окончательно установленным, является открытие Аберсоном в растениях сем. Crassulaceae яблочной кислоты НО—СО—ОН(ОН)—СН2—СО—ОН, отличной от обыкновенной, ее антипода и их рацемического полимера. Для тела с одним асимметричным атомом углерода другие изомеры при свободе вращения углеродных систем немыслимы], то должно допустить, что тетраэдры, находящиеся в положении фиг. 7, обладают свободным вращением вокруг оси, их соединяющей; все формулы изомерных глюкоз и их производных, рассмотренные ранее, выведены именно в предположении такого свободного вращения. Совершенно противоположное отношение приходится принять для двух атомов углерода, соединенных двойною связью, т. е. для производных этилена формулы abC:Ccd, где а, b, с и d представляют любые одноатомные остатки (атомы). С точки зрения тетраэдрической гипотезы (Вант-Гоффа) соединение двух атомов углерода двойною связью приходится изображать двумя тетраэдрами, соединенными уже не вершинами, а ребром (фиг. 11), и Вант-Гофф с самого начала принял, что в таких системах относительное положение групп, соединенных с углеродом, фиксировано и что они расположены в одной плоскости. Фиг. 11. Фиг. 12. Фиг. 13Это последнее обстоятельство уже заранее решает, что при четырех различных заместителях для этилена невозможны асимметрические, энантиоморфные конфигурации, и тем самым устраняет возможность для них оптической изомерии (см. Стереохимия) [Ле-Бель первоначально предполагал, что группу )С=С( можно будет в некоторых случаях приравнять одному С, что заставляло ожидать для C2abcd оптических изомеров]. Это же допущение Вант-Гоффа ведет к предвидению для тел структурной формулы abC=Cab двух геометрических изомеров (фиг. 12 и 13), которые схематически можно передать формулами:
Фиг. 11. Фиг. 12. Фиг. 13Это последнее обстоятельство уже заранее решает, что при четырех различных заместителях для этилена невозможны асимметрические, энантиоморфные конфигурации, и тем самым устраняет возможность для них оптической изомерии (см. Стереохимия) [Ле-Бель первоначально предполагал, что группу )С=С( можно будет в некоторых случаях приравнять одному С, что заставляло ожидать для C2abcd оптических изомеров]. Это же допущение Вант-Гоффа ведет к предвидению для тел структурной формулы abC=Cab двух геометрических изомеров (фиг. 12 и 13), которые схематически можно передать формулами: И то и другое дальнейшими опытами подтвердилось вполне. Во-первых, все попытки разделения на оптические изомеры тел формулы C2abcd и C2a2b2 потерпели полную неудачу (Ле-Бель; Кекуле и Аншютц). Одно время Ле-Бель думал, что культивированием плесени в растворах цитраконовой и мезаконовой НО—ОС—СН=С(СН3)—СО—ОН кислот ему удалось получить искомые оптически деятельные вещества, но ближайшее исследование показало, что в данном случае фермент плесени является причиною реакции гидратации, ведущей к образованию d- (из мезаконовой) и l- (из цитраконовой) метиляблочных кислот (Бишоф, Ле-Бель).
И то и другое дальнейшими опытами подтвердилось вполне. Во-первых, все попытки разделения на оптические изомеры тел формулы C2abcd и C2a2b2 потерпели полную неудачу (Ле-Бель; Кекуле и Аншютц). Одно время Ле-Бель думал, что культивированием плесени в растворах цитраконовой и мезаконовой НО—ОС—СН=С(СН3)—СО—ОН кислот ему удалось получить искомые оптически деятельные вещества, но ближайшее исследование показало, что в данном случае фермент плесени является причиною реакции гидратации, ведущей к образованию d- (из мезаконовой) и l- (из цитраконовой) метиляблочных кислот (Бишоф, Ле-Бель). Безрезультатны были и попытки активирования аллилового спирта СН2=СН—СН2—ОН и твердой кротоновой кислоты СН3—СН=СН—СО—ОН; они не приобрели вращательной способности, и Ле-Бель заканчивает свою статью по этому вопросу словами: "на основании этих исследований можно прийти к заключению, что первые продукты замещения этилена имеют действительно ту плоскую форму, которая им была приписана (Вант Гоффом)". Во-вторых — ко времени появления стереохимической гипотезы между непредельными соединениями существовало много изомеров, обладавших различными физическими свойствами, различной кристаллической формой (при отсутствии энантиоморфизма), различными: растворимостью, удельными весами, температурами плавления и кипения и отчасти отличными химическими свойствами, но которым, тем не менее, приходилось приписать (по реакциям образования, напр.) большею частью тождественные структурные формулы; таковы, напр., были кислоты: малеиновая и фумаровая НООС—СН=СН—СООН, кротоновая (см.) и изокротоновая C4H6O2, ангеликовая и тиглиновая — С3Н8О2 (см.) и т. д.; иногда таким изомерам, вопреки большинству известных для них реакций, приписывались различные формулы; считалось, напр., возможным, что формула малеиновой кислоты
Безрезультатны были и попытки активирования аллилового спирта СН2=СН—СН2—ОН и твердой кротоновой кислоты СН3—СН=СН—СО—ОН; они не приобрели вращательной способности, и Ле-Бель заканчивает свою статью по этому вопросу словами: "на основании этих исследований можно прийти к заключению, что первые продукты замещения этилена имеют действительно ту плоскую форму, которая им была приписана (Вант Гоффом)". Во-вторых — ко времени появления стереохимической гипотезы между непредельными соединениями существовало много изомеров, обладавших различными физическими свойствами, различной кристаллической формой (при отсутствии энантиоморфизма), различными: растворимостью, удельными весами, температурами плавления и кипения и отчасти отличными химическими свойствами, но которым, тем не менее, приходилось приписать (по реакциям образования, напр.) большею частью тождественные структурные формулы; таковы, напр., были кислоты: малеиновая и фумаровая НООС—СН=СН—СООН, кротоновая (см.) и изокротоновая C4H6O2, ангеликовая и тиглиновая — С3Н8О2 (см.) и т. д.; иногда таким изомерам, вопреки большинству известных для них реакций, приписывались различные формулы; считалось, напр., возможным, что формула малеиновой кислоты , а фумаровой — приведенная выше и т. д. Теория Вант-Гоффа объяснила такие случаи и дала им рациональные формулы, а найденные затем факты подтвердили правильность последних. Особенно удачным можно считать подтверждение формул малеиновой и фумаровой кислот. Для первой Вант-Гофф, руководствуясь ее легкой способностью терять элементы воды с образованием ангидрида, дал формулу
, а фумаровой — приведенная выше и т. д. Теория Вант-Гоффа объяснила такие случаи и дала им рациональные формулы, а найденные затем факты подтвердили правильность последних. Особенно удачным можно считать подтверждение формул малеиновой и фумаровой кислот. Для первой Вант-Гофф, руководствуясь ее легкой способностью терять элементы воды с образованием ангидрида, дал формулу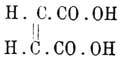 ,в которой карбоксильные группы возможно сближены, а для второй была предложена формула
,в которой карбоксильные группы возможно сближены, а для второй была предложена формула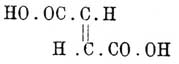 и сначала Танатар, а затем Кекуле и Аншютц, получили при окислении их натровых солей слабым раствором марганцево-калиевой соли из малеиновой кислоты — мезовинную, а из фумаровой — виноградную. Реакция протекает буквально так, как следовало ожидать на основании стереохимических формул; действительно, сводится она на присоединение по месту двойной связи двух гидроксильных групп; на счет какой бы из связей этот процесс ни шел, мы должны ожидать для вещества с формулой
и сначала Танатар, а затем Кекуле и Аншютц, получили при окислении их натровых солей слабым раствором марганцево-калиевой соли из малеиновой кислоты — мезовинную, а из фумаровой — виноградную. Реакция протекает буквально так, как следовало ожидать на основании стереохимических формул; действительно, сводится она на присоединение по месту двойной связи двух гидроксильных групп; на счет какой бы из связей этот процесс ни шел, мы должны ожидать для вещества с формулой образования кислоты
образования кислоты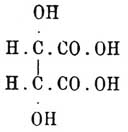 ,хотя и содержащей два асимметричных атома углерода, но такую, обе половины которой могут быть рассматриваемы, как зеркальные изображения друг друга, а это и есть мезовинная кислота (см. Винная), наоборот, для
,хотя и содержащей два асимметричных атома углерода, но такую, обе половины которой могут быть рассматриваемы, как зеркальные изображения друг друга, а это и есть мезовинная кислота (см. Винная), наоборот, для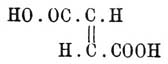 в зависимости от того, на счет какой из связей двойной связи произойдет присоединение гидроксилов, а разрыв той или другой (ввиду их равноценности) одинаково вероятен, должны получиться две энантиоморфных формы:
в зависимости от того, на счет какой из связей двойной связи произойдет присоединение гидроксилов, а разрыв той или другой (ввиду их равноценности) одинаково вероятен, должны получиться две энантиоморфных формы: а это и суть формулы (по Э. Фишеру) правой и левой винной кислот (см. выше), соединение же их и есть виноградная кислота. Таким образом, число изомеров, их химический характер оказываются отвечающими предвидениям теории. Как подтверждение ее должно рассматривать и то обстоятельство, что при окислении циклических тел, стереометрическое положение групп в которых тоже должно считаться фиксированным, получаются производные малеиновой кислоты. Так, Кекуле и Штрекер получили при окислении бензола хлорноватистой кислотой — трихлорацетилакриловую кисл. CaCl3O—CH=СН—СО—ОН, которая под влиянием щелочей дает хлороформ и малеиновую, а не фумаровую [Кариус ранее получил при этой реакции фумаровую; результат, по Кекуле, объясняется изомеризующим действием кислот, на которое Кариус не обратил внимания]:
а это и суть формулы (по Э. Фишеру) правой и левой винной кислот (см. выше), соединение же их и есть виноградная кислота. Таким образом, число изомеров, их химический характер оказываются отвечающими предвидениям теории. Как подтверждение ее должно рассматривать и то обстоятельство, что при окислении циклических тел, стереометрическое положение групп в которых тоже должно считаться фиксированным, получаются производные малеиновой кислоты. Так, Кекуле и Штрекер получили при окислении бензола хлорноватистой кислотой — трихлорацетилакриловую кисл. CaCl3O—CH=СН—СО—ОН, которая под влиянием щелочей дает хлороформ и малеиновую, а не фумаровую [Кариус ранее получил при этой реакции фумаровую; результат, по Кекуле, объясняется изомеризующим действием кислот, на которое Кариус не обратил внимания]: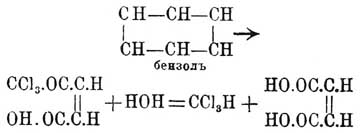 Дёбнер наблюдал при окислении фенола марганцево-калиевой солью образование щавелевой и мезовинной кислот (см.); последняя, по всей вероятности, является продуктом окисления малеиновой кислоты. Цинке при действии хлорноватистой щелочи на параамидофенол получил дихлормалеиновую, а на резорцин — дихлорацетилтрихлорокротоновую кислоту.
Дёбнер наблюдал при окислении фенола марганцево-калиевой солью образование щавелевой и мезовинной кислот (см.); последняя, по всей вероятности, является продуктом окисления малеиновой кислоты. Цинке при действии хлорноватистой щелочи на параамидофенол получил дихлормалеиновую, а на резорцин — дихлорацетилтрихлорокротоновую кислоту. Наконец, Чиамичиан превратил целый ряд производных пиррола, фурфурола и тиофена в производные малеиновой кислоты, а их β-метилированные производные — в производные цитраконовой кислоты; так, из четырехбромистого тиофена
Наконец, Чиамичиан превратил целый ряд производных пиррола, фурфурола и тиофена в производные малеиновой кислоты, а их β-метилированные производные — в производные цитраконовой кислоты; так, из четырехбромистого тиофена он получил дибромомалеиновую кислоту
он получил дибромомалеиновую кислоту 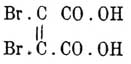 , а из трехбромистого тиотолена
, а из трехбромистого тиотолена  — бромоцитраконовую кислоту:
— бромоцитраконовую кислоту:  . В отличие от предельных соединений установление для замещенных этиленов стереохимической формулы очень затруднительно. Причиною является сравнительно большая легкость взаимных превращений стереоизомеров. Так, малеиновая кислота при осторожном нагревании до 150° может быть почти количественно превращена в фумаровую [Превращение идет с выделением тепла, следовательно, отношение между малеиновой и фумаровой кислотами напоминает отношение переохлажденной жидкости к отвечающему ей твердому телу], изокротоновая жидкая при повторенных перегонках переходит в кротоновую кислоту твердую (см.) и т. д. Очень часто такие превращения возбуждаются каталитическими телами. Так, по Петри, бром и некоторые минеральные кислоты превращают малеиновую кислоту в фумаровую; то же наблюдено Фиттигом и Вислиценусом для ангеликовой и тиглиновой кислот, причем Фиттиг еще подметил ускоряющее действие света на превращение ангеликовой кислоты в присутствии брома [Превращается ли ангеликововая кислота под влиянием света в тиглиновую в отсутствие брома — неизвестно, но этого должно ожидать]. Соли малеиновой кислоты превращаются в соли фумаровой (Скрауп); следы йода (Аншютц) вызывают превращение эфиров малеиновой кислоты в эфиры фумаровой; ничтожные количества окислов азота превращают кислоты: олеиновую, гипогеиновую и эруковую в их стереоизомеры — кислоты: элаидиновую, гаидиновую и брассидиновую по схеме:
. В отличие от предельных соединений установление для замещенных этиленов стереохимической формулы очень затруднительно. Причиною является сравнительно большая легкость взаимных превращений стереоизомеров. Так, малеиновая кислота при осторожном нагревании до 150° может быть почти количественно превращена в фумаровую [Превращение идет с выделением тепла, следовательно, отношение между малеиновой и фумаровой кислотами напоминает отношение переохлажденной жидкости к отвечающему ей твердому телу], изокротоновая жидкая при повторенных перегонках переходит в кротоновую кислоту твердую (см.) и т. д. Очень часто такие превращения возбуждаются каталитическими телами. Так, по Петри, бром и некоторые минеральные кислоты превращают малеиновую кислоту в фумаровую; то же наблюдено Фиттигом и Вислиценусом для ангеликовой и тиглиновой кислот, причем Фиттиг еще подметил ускоряющее действие света на превращение ангеликовой кислоты в присутствии брома [Превращается ли ангеликововая кислота под влиянием света в тиглиновую в отсутствие брома — неизвестно, но этого должно ожидать]. Соли малеиновой кислоты превращаются в соли фумаровой (Скрауп); следы йода (Аншютц) вызывают превращение эфиров малеиновой кислоты в эфиры фумаровой; ничтожные количества окислов азота превращают кислоты: олеиновую, гипогеиновую и эруковую в их стереоизомеры — кислоты: элаидиновую, гаидиновую и брассидиновую по схеме:![По Аншютцу, кислоты малеинаниловая и малеинаминовая превращаются при кипячении со щелочью в фумаровую кислоту, а по Делилю, цитраконовая в тех же условиях переходит в мезаконовую [В этом направлении собран обширный материал Фиттигом с учениками.] и т. д. О воззрениях Вислиценуса на подобные превращения см.](/pictures/brokgauz_efron/b62_618-9.jpg) По Аншютцу, кислоты малеинаниловая и малеинаминовая превращаются при кипячении со щелочью в фумаровую кислоту, а по Делилю, цитраконовая в тех же условиях переходит в мезаконовую [В этом направлении собран обширный материал Фиттигом с учениками.] и т. д. О воззрениях Вислиценуса на подобные превращения см. "Abhandl. d. mathem., phys. Class. der Sächs. Gesel. der Wissensch.", т. XIV, или подробные руководства по стереохимии.Геометрическая изомерия известна и для предельных соединений; по крайней мере, под названием геометрических изомеров описаны многочисленные изомеры двузамещенных янтарных кислот (см. Янтарная кисл. и ее гомологи) HO—OC—CHX—CHY—CO—OH [Х и Y одноатомные группы], полученные в последние года Ауверсом, В. Мейером, Зелинским, Бишофом и др. В большинстве случаев для данных Х и Y наблюдается пара изомеров, из которых один плавится при сравнительно низкой температуре, более растворим и легче превращается в ангидрид, теряя элементы воды; другой — плавится выше, растворим труднее, дает меньше ангидрида в условиях, тождественных с первым изомером; оба способны в известных условиях переходить друг в друга (подробн. см. Янтарные кисл.). Так как эти отношения очень близки в тому, что известно для пары геометрически изомерных малеиновой и фумаровой кислот, то и двузамещенные янтарные кислоты получили названия — низкоплавящиеся и т. д. — малеиноидных, а высокоплавящиеся — фумароидных. По мнению многих (особенно Бишофа), изомерия этих кислот должна объясняться относительной фиксацией в пространстве групп, соединенных с двумя средними асимметричными атомами углерода; расположение этих групп (по Байеру) в малеиноидных кислотах
По Аншютцу, кислоты малеинаниловая и малеинаминовая превращаются при кипячении со щелочью в фумаровую кислоту, а по Делилю, цитраконовая в тех же условиях переходит в мезаконовую [В этом направлении собран обширный материал Фиттигом с учениками.] и т. д. О воззрениях Вислиценуса на подобные превращения см. "Abhandl. d. mathem., phys. Class. der Sächs. Gesel. der Wissensch.", т. XIV, или подробные руководства по стереохимии.Геометрическая изомерия известна и для предельных соединений; по крайней мере, под названием геометрических изомеров описаны многочисленные изомеры двузамещенных янтарных кислот (см. Янтарная кисл. и ее гомологи) HO—OC—CHX—CHY—CO—OH [Х и Y одноатомные группы], полученные в последние года Ауверсом, В. Мейером, Зелинским, Бишофом и др. В большинстве случаев для данных Х и Y наблюдается пара изомеров, из которых один плавится при сравнительно низкой температуре, более растворим и легче превращается в ангидрид, теряя элементы воды; другой — плавится выше, растворим труднее, дает меньше ангидрида в условиях, тождественных с первым изомером; оба способны в известных условиях переходить друг в друга (подробн. см. Янтарные кисл.). Так как эти отношения очень близки в тому, что известно для пары геометрически изомерных малеиновой и фумаровой кислот, то и двузамещенные янтарные кислоты получили названия — низкоплавящиеся и т. д. — малеиноидных, а высокоплавящиеся — фумароидных. По мнению многих (особенно Бишофа), изомерия этих кислот должна объясняться относительной фиксацией в пространстве групп, соединенных с двумя средними асимметричными атомами углерода; расположение этих групп (по Байеру) в малеиноидных кислотах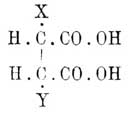 т. е. они отвечают мезовинной кислоте, в которой водные остатки замещены Х и Y; то же самое Бишоф выражает стереометрической формулой, данной фиг. 14, которая представляет не что иное, как фиг. 7, рассматриваемую сверху; по аналогии с мезовинной — антивинной Бишоф называет малеиноидные кислоты еще антикислотами. Кислоты фумароидные, по Байеру, отвечают виноградной, что и выражается им формулой:
т. е. они отвечают мезовинной кислоте, в которой водные остатки замещены Х и Y; то же самое Бишоф выражает стереометрической формулой, данной фиг. 14, которая представляет не что иное, как фиг. 7, рассматриваемую сверху; по аналогии с мезовинной — антивинной Бишоф называет малеиноидные кислоты еще антикислотами. Кислоты фумароидные, по Байеру, отвечают виноградной, что и выражается им формулой: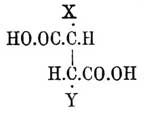 Бишоф называет их с тою же целью паракислотами (от паравинной) и выражает их строение фиг. 15.
Бишоф называет их с тою же целью паракислотами (от паравинной) и выражает их строение фиг. 15.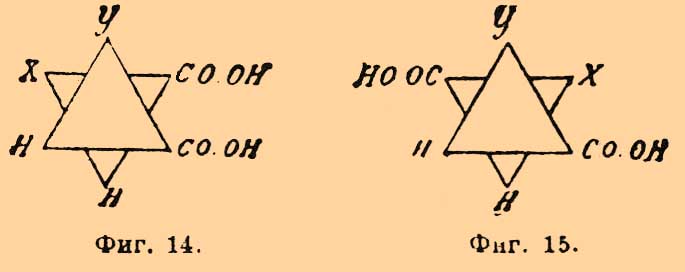 Фиг. 14. Фиг. 15Эти соображения требуют в настоящее время полного пересмотра. Начать с того, что нет никакой внутренней необходимости в том, чтобы кислоты мезовинного типа плавились всегда ниже кислот виноградного типа, а потому неосновательно считать нижеплавящиеся кислоты всегда за антиизомеры; еще важнее то обстоятельство, что только для общей формулы НО—ОС—СНХ—СНХ—СО—ОН, делимой пополам, мыслимы оптически недеятельные изомеры мезовинного типа; когда же мы имеем разные заместители, например HO—OC—CHX—CHY—CO—OH, то формула не состоит из тождественных половин и тогда, как известно, N = 2n, Na = 2n, Nr = 2n—1 и Ni = 0, т. е. тогда возможны только тела виноградного типа, иначе — только паракислоты (ср. что сказано о дибромкоричной и дибромаллокоричной кислотах). Нельзя не обратить, кроме того, внимания на то, что до сих пор все попытки деления на оптические антиподы двузамещенных янтарных паракислот оказались безрезультатными. На почве гипотезы тетраэдричного расположения групп, соединенных с углеродными атомами, мы можем составить себе некоторое представление об относительных расстояниях атомов в молекуле и, оказывается, соображения этого рода позволяют легко объяснить некоторые давно уже известные факты. Если довольствоваться только структурною формулой, например пентана:
Фиг. 14. Фиг. 15Эти соображения требуют в настоящее время полного пересмотра. Начать с того, что нет никакой внутренней необходимости в том, чтобы кислоты мезовинного типа плавились всегда ниже кислот виноградного типа, а потому неосновательно считать нижеплавящиеся кислоты всегда за антиизомеры; еще важнее то обстоятельство, что только для общей формулы НО—ОС—СНХ—СНХ—СО—ОН, делимой пополам, мыслимы оптически недеятельные изомеры мезовинного типа; когда же мы имеем разные заместители, например HO—OC—CHX—CHY—CO—OH, то формула не состоит из тождественных половин и тогда, как известно, N = 2n, Na = 2n, Nr = 2n—1 и Ni = 0, т. е. тогда возможны только тела виноградного типа, иначе — только паракислоты (ср. что сказано о дибромкоричной и дибромаллокоричной кислотах). Нельзя не обратить, кроме того, внимания на то, что до сих пор все попытки деления на оптические антиподы двузамещенных янтарных паракислот оказались безрезультатными. На почве гипотезы тетраэдричного расположения групп, соединенных с углеродными атомами, мы можем составить себе некоторое представление об относительных расстояниях атомов в молекуле и, оказывается, соображения этого рода позволяют легко объяснить некоторые давно уже известные факты. Если довольствоваться только структурною формулой, например пентана: , то можно думать, что углерод 1-й ближе всего к углероду 2-му и наиболее удален от углерода 5-го, а стереохимическая модель (фиг. 16; изображены только атомы углерода, или вернее, сферы действия их сродств) позволяет ожидать, что группа а, связанная с первым углеродом, всего легче вступит во взаимодействие с группою, связанной с 5-м атомом углерода, и всего труднее с группою, связанною со 2-м атомом углерода.
, то можно думать, что углерод 1-й ближе всего к углероду 2-му и наиболее удален от углерода 5-го, а стереохимическая модель (фиг. 16; изображены только атомы углерода, или вернее, сферы действия их сродств) позволяет ожидать, что группа а, связанная с первым углеродом, всего легче вступит во взаимодействие с группою, связанной с 5-м атомом углерода, и всего труднее с группою, связанною со 2-м атомом углерода. Фиг. 16.Опытный материал вполне гармонирует с таким заключением. Давно было известно, что внутримолекулярные реакции очень трудно идут между группами сосдних (α) углеродных атомов, чаще наблюдаются они для β-положения (1,3) и еще чаще для γ- (1,4) и δ- (1,5) положений. Так, β-лактоны (см.) почти неизвестны, а существуют предпочтительно γ- и δ-лактоны; α- и β-амидокислоты: HO—OC—CHNH2—R и HO—OC—CH2—CHNH2—R не теряют воды, a γ- — HO—OC—[CH2]2—CHNH2R и δ- — HO—OC—[CH2]3—CHNH2—R легко образуют воду и соответственные лактамы: пирримидоны и пиперидоны (см.). Ни щавелевая кисл., ни малоновая не дают ангидридов, каковые получаются для янтарной кислоты и ее гомологов (ср. выше) и для глутаровых кислот [Явление находится в зависимости, кроме того, от степени замещения кислоты; многозамещенные образуют ангидриды легче (Бишоф, Гьельт)]. К этой же области явлений должна быть отнесена и легкая способность γ-дикетонов давать производные фурфурана, пиролла и тиофена:
Фиг. 16.Опытный материал вполне гармонирует с таким заключением. Давно было известно, что внутримолекулярные реакции очень трудно идут между группами сосдних (α) углеродных атомов, чаще наблюдаются они для β-положения (1,3) и еще чаще для γ- (1,4) и δ- (1,5) положений. Так, β-лактоны (см.) почти неизвестны, а существуют предпочтительно γ- и δ-лактоны; α- и β-амидокислоты: HO—OC—CHNH2—R и HO—OC—CH2—CHNH2—R не теряют воды, a γ- — HO—OC—[CH2]2—CHNH2R и δ- — HO—OC—[CH2]3—CHNH2—R легко образуют воду и соответственные лактамы: пирримидоны и пиперидоны (см.). Ни щавелевая кисл., ни малоновая не дают ангидридов, каковые получаются для янтарной кислоты и ее гомологов (ср. выше) и для глутаровых кислот [Явление находится в зависимости, кроме того, от степени замещения кислоты; многозамещенные образуют ангидриды легче (Бишоф, Гьельт)]. К этой же области явлений должна быть отнесена и легкая способность γ-дикетонов давать производные фурфурана, пиролла и тиофена: Одно из следствий теории до сих пор, впрочем, не подтверждено. Если строить формулы предельных одноосновных кислот из моделей, то нетрудно видеть, что начиная с кислот, заключающих C6 и более в молекуле, углеродные атомы уже не находятся в одной плоскости, а образуют довольно сложную фигуру, которая может быть асимметричной, не совместимой с ее зеркальным изображением; следовало бы потому ожидать для высших одноосновных жирных кислот оптической деятельности; таковая пока не известна.С. ацетилена и циклических соединений. При тетраэдрической гипотезе тройную связь необходимо представить себе в виде соприкосновения двух тетраэдров по одной плоскости; стереохимическая формула ацетилена потому передается фиг. 17; как видно — в ацетилене все атомы находятся не только в одной плоскости (это имеется и в этилене), но и на одной прямой линии.
Одно из следствий теории до сих пор, впрочем, не подтверждено. Если строить формулы предельных одноосновных кислот из моделей, то нетрудно видеть, что начиная с кислот, заключающих C6 и более в молекуле, углеродные атомы уже не находятся в одной плоскости, а образуют довольно сложную фигуру, которая может быть асимметричной, не совместимой с ее зеркальным изображением; следовало бы потому ожидать для высших одноосновных жирных кислот оптической деятельности; таковая пока не известна.С. ацетилена и циклических соединений. При тетраэдрической гипотезе тройную связь необходимо представить себе в виде соприкосновения двух тетраэдров по одной плоскости; стереохимическая формула ацетилена потому передается фиг. 17; как видно — в ацетилене все атомы находятся не только в одной плоскости (это имеется и в этилене), но и на одной прямой линии.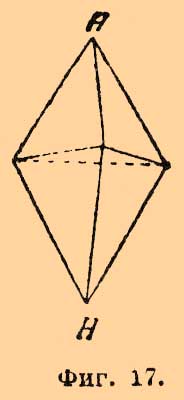 Фиг. 17.Характер строения замещенных ацетиленов находится в полной зависимости от строения замещающих групп и потому сводится на изложенное выше [Миную гипотезу Байера о натяжениях сродств в ацетилене и циклических производных: она не находится в связи с тем, что изложено в настоящей статье. Для желающих перечисляю в конце литературу предмета]. Переходя к С. циклических тел, я считаю возможным остановиться только на трех обстоятельствах. 1) Формула бензола, являющегося наиболее хорошо изученным циклическим телом, не может быть выражена призмой, потому что при призматическом строении уже двузамещенные продукты его должны были бы, как энантиаморфные, обладать оптической деятельностью. На это обстоятельство впервые обратил внимание Вант-Гофф. Действительно, одного взгляда на фиг. 18 и 19 достаточно, чтобы видеть, что такие построения не совместимы, а потому салициловая кисл., ее альдегиды, ортотолуидин и т. д. должны бы существовать в двух оптически деятельных видоизменениях, отвечающих правой и левой винным кислотам; ничего подобного, однако, не известно, и Ле-Бель тщетно пытался разделить обыкновенный ортотолуидин, считая его за рацемическое тело, на оптические антиподы.
Фиг. 17.Характер строения замещенных ацетиленов находится в полной зависимости от строения замещающих групп и потому сводится на изложенное выше [Миную гипотезу Байера о натяжениях сродств в ацетилене и циклических производных: она не находится в связи с тем, что изложено в настоящей статье. Для желающих перечисляю в конце литературу предмета]. Переходя к С. циклических тел, я считаю возможным остановиться только на трех обстоятельствах. 1) Формула бензола, являющегося наиболее хорошо изученным циклическим телом, не может быть выражена призмой, потому что при призматическом строении уже двузамещенные продукты его должны были бы, как энантиаморфные, обладать оптической деятельностью. На это обстоятельство впервые обратил внимание Вант-Гофф. Действительно, одного взгляда на фиг. 18 и 19 достаточно, чтобы видеть, что такие построения не совместимы, а потому салициловая кисл., ее альдегиды, ортотолуидин и т. д. должны бы существовать в двух оптически деятельных видоизменениях, отвечающих правой и левой винным кислотам; ничего подобного, однако, не известно, и Ле-Бель тщетно пытался разделить обыкновенный ортотолуидин, считая его за рацемическое тело, на оптические антиподы.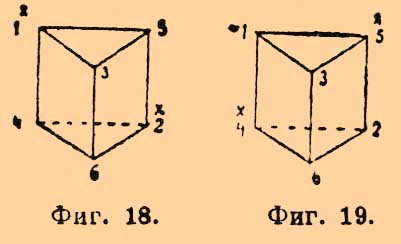 Фиг. 18. Фиг. 19.Выводом из этого является представление, что в бензоле (и в других циклических соединениях) атомы углерода лежат или в одной, или в двух параллельных плоскостях; при таком допущении атомы водорода углеводородов или замещающие их группы тоже могут находиться в плоскостях, параллельных первым [Это видно на модели. Длинные боковые цепи занимают, понятно, им свойственное положение]. 2) При распределении атомов в различных параллельных плоскостях для различных циклических производных возможна изомерия, вполне подобная той, которая известна для винных кислот. Так, принимая, что шесть С атомов гексагидробензола лежат в одной плоскости, мы для гексагидрофталевой кислоты можем ожидать, что оба карбоксила ее будут приходиться с одной стороны этой плоскости, и тогда ей можно придать такую формулу:
Фиг. 18. Фиг. 19.Выводом из этого является представление, что в бензоле (и в других циклических соединениях) атомы углерода лежат или в одной, или в двух параллельных плоскостях; при таком допущении атомы водорода углеводородов или замещающие их группы тоже могут находиться в плоскостях, параллельных первым [Это видно на модели. Длинные боковые цепи занимают, понятно, им свойственное положение]. 2) При распределении атомов в различных параллельных плоскостях для различных циклических производных возможна изомерия, вполне подобная той, которая известна для винных кислот. Так, принимая, что шесть С атомов гексагидробензола лежат в одной плоскости, мы для гексагидрофталевой кислоты можем ожидать, что оба карбоксила ее будут приходиться с одной стороны этой плоскости, и тогда ей можно придать такую формулу: (оба карбоксила находятся в плоскости, параллельной плоскости бумаги, на которой, предполагается, лежит кольцо из 6 С); подобные формулы Байер называет cis-формулами; с одинаковым правом такие соединения можно бы назвать антисоединениями; построение, им отвечающее, вполне симметрично; это легко видеть, если представить себе, что перпендикулярно к плоскости бумаги помещено зеркало ab, обозначенное пунктирной линией: обе половины формулы явятся тогда зеркальным изображением друг друга, а следовательно, тело, им отвечающее, несмотря на содержание двух асимметричных атомов углерода, должно быть оптически недеятельным — типа мезовинной кисл. Можно, однако, для гексагидрофталевой кислоты расположить карбоксилы и иначе: один под плоскостью, содержащей 6 С бензольного кольца, и другой над этой плоскостью:
(оба карбоксила находятся в плоскости, параллельной плоскости бумаги, на которой, предполагается, лежит кольцо из 6 С); подобные формулы Байер называет cis-формулами; с одинаковым правом такие соединения можно бы назвать антисоединениями; построение, им отвечающее, вполне симметрично; это легко видеть, если представить себе, что перпендикулярно к плоскости бумаги помещено зеркало ab, обозначенное пунктирной линией: обе половины формулы явятся тогда зеркальным изображением друг друга, а следовательно, тело, им отвечающее, несмотря на содержание двух асимметричных атомов углерода, должно быть оптически недеятельным — типа мезовинной кисл. Можно, однако, для гексагидрофталевой кислоты расположить карбоксилы и иначе: один под плоскостью, содержащей 6 С бензольного кольца, и другой над этой плоскостью: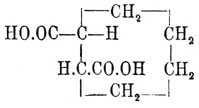 такое построение энантиоморфно асимметрично, и оно должно отвечать правой и левой винным кислотам; Байер называет такое тело trans-изомером: можно назвать его и параизомером. Случаи установленной cis- и trans-изомерии настолько многочисленны, что перечислить их в настоящей статье нет возможности. Достаточно сказать, что они наблюдаются не только для гидрогенизованных ароматических производных, но и для производных три-, тетра- и пентаметиленов (и будут найдены и для гепта-, окто- и т. п. метиленов), для пиперазонов, тритиоальдегидов и т. д. 3) Возможно, что cis-trans-изомерией необходимо объяснить и случаи оптической деятельности таких тел, которые, судя по структурной формуле, таковой не должны бы обладать. Сюда, напр., относится оптическая деятельность инозита — гексаоксигексагидробензола
такое построение энантиоморфно асимметрично, и оно должно отвечать правой и левой винным кислотам; Байер называет такое тело trans-изомером: можно назвать его и параизомером. Случаи установленной cis- и trans-изомерии настолько многочисленны, что перечислить их в настоящей статье нет возможности. Достаточно сказать, что они наблюдаются не только для гидрогенизованных ароматических производных, но и для производных три-, тетра- и пентаметиленов (и будут найдены и для гепта-, окто- и т. п. метиленов), для пиперазонов, тритиоальдегидов и т. д. 3) Возможно, что cis-trans-изомерией необходимо объяснить и случаи оптической деятельности таких тел, которые, судя по структурной формуле, таковой не должны бы обладать. Сюда, напр., относится оптическая деятельность инозита — гексаоксигексагидробензола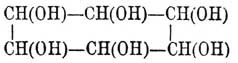 (см. Гидраты углерода),хинной кислоты и т. п. Пример инозита и мезовинной кислоты показывают, между прочим, что неправильно Вант-Гоффом оптическая деятельность сведена только к асимметрии углерода, так как цепь углеродных атомов (будет ли она закрытая или открытая) сама по себе почти всегда совместима с собою [Несовместимость мыслима только при винтообразном расположении атомов С.: правые и левые винты энантиоморфны]; не совместимо же, энантиаморфно — может быть расположение в пространстве атомов, соединенных с углеродом или другим поливалентным атомом. О геометрической изомерии углеродоазотных соединений см. Изонитрозосоединения; о геометрической С. азота см. Диазосоединения, а также специальную литературу.Литература. J. H. van't Hoff — F. Herrmann, "Die Lagerung der Atome im Raum" (1877); A. Wunderlich, "Configuration organischer Molekule" (1886); J. H. van't Hoff, "Dix années dans l'histoire d'une théorie" (1887; перепечатана здесь и первая ст. Le-Веl'я по стереохимии); J. Wislicenus, "Ueber die räumliche Anordnung der Atome in organischen Molekulen etc." (в "Abhandl. der mat.-phys. Classe der K. Sächs. Gesellsch. der Wissensch.", XIV, 1—77; реферат части ст. в "Журн. Русск. хим. общ.", т. XX, 1888 [2] 1—21); К. Auwers, "Die Entwicklung der Stereochemie" (1890); Ph. A. Guye, "Etude sur la dissimétrie moléculaire" (1891); G. Errera, "Lezioni sulla polarimetria" (1891);. Ш. M. Безредка (под ред. Н. Д. Зелинского), "Опыт истории развития стереохимических представлений" (1892); van't Hoff — W. Meyerhoffer, "Stereochemie" (1892); Ch. Friedel-Benzène (constitution, 2-ème Suppl. au Dictionnaire etc. de Wurtz, 1-ère partie, p. 451, 1892); J. Wislicenus, "Die Chemie und das Problem von der Materie" (1893); A. Hantzsch, "Grundriss der Stereochemie" (1893); J. H. van't Hoff, "Die Lagerung der Atome im Raume" (2 изд., 1894); С. A. Bischoff вместе с Р. Walden, "Handbuch der Stereochemie" (1894); А. Hantzsch, "Précis de Stéréochimie" (1896); H. Landolt, "Das optische Drehungsvermögen" (2 изд., 1898); Н. Вальден, "Материалы к изучению оптической изомерии" (1898: отдельными главами напечатано в т. XXX "Ж. Р. ф.-хим. общ.", стр. 483, 491, 502, 632 и 767); J. H. van't Hoff, "Vorlesungen ü. theoret. u. phys. Chemie" (2 тетр., "Die chemische Statik", стр. 94—123, 1899); P. Freundler, "La Stéréochimie" ("Scientia", № 3, 1899). Рефераты С. A. Bischoff'a в Meyer's "Jahrbuch der Chemie", отд. "Organische Chemie" — "Stereoisomerie" с 1891 г., и П. Вальден, "Оптическая изомерия" ("Ж. Р. ф.-х. о.", т. XXXII, [2], 42—59); многочисл. отд. статьи в спец. хим. журн.; E. Fischer'a в "Ber. d. deutsch. Chem. Gesellschaft".A. И. Горбов. Δ.
(см. Гидраты углерода),хинной кислоты и т. п. Пример инозита и мезовинной кислоты показывают, между прочим, что неправильно Вант-Гоффом оптическая деятельность сведена только к асимметрии углерода, так как цепь углеродных атомов (будет ли она закрытая или открытая) сама по себе почти всегда совместима с собою [Несовместимость мыслима только при винтообразном расположении атомов С.: правые и левые винты энантиоморфны]; не совместимо же, энантиаморфно — может быть расположение в пространстве атомов, соединенных с углеродом или другим поливалентным атомом. О геометрической изомерии углеродоазотных соединений см. Изонитрозосоединения; о геометрической С. азота см. Диазосоединения, а также специальную литературу.Литература. J. H. van't Hoff — F. Herrmann, "Die Lagerung der Atome im Raum" (1877); A. Wunderlich, "Configuration organischer Molekule" (1886); J. H. van't Hoff, "Dix années dans l'histoire d'une théorie" (1887; перепечатана здесь и первая ст. Le-Веl'я по стереохимии); J. Wislicenus, "Ueber die räumliche Anordnung der Atome in organischen Molekulen etc." (в "Abhandl. der mat.-phys. Classe der K. Sächs. Gesellsch. der Wissensch.", XIV, 1—77; реферат части ст. в "Журн. Русск. хим. общ.", т. XX, 1888 [2] 1—21); К. Auwers, "Die Entwicklung der Stereochemie" (1890); Ph. A. Guye, "Etude sur la dissimétrie moléculaire" (1891); G. Errera, "Lezioni sulla polarimetria" (1891);. Ш. M. Безредка (под ред. Н. Д. Зелинского), "Опыт истории развития стереохимических представлений" (1892); van't Hoff — W. Meyerhoffer, "Stereochemie" (1892); Ch. Friedel-Benzène (constitution, 2-ème Suppl. au Dictionnaire etc. de Wurtz, 1-ère partie, p. 451, 1892); J. Wislicenus, "Die Chemie und das Problem von der Materie" (1893); A. Hantzsch, "Grundriss der Stereochemie" (1893); J. H. van't Hoff, "Die Lagerung der Atome im Raume" (2 изд., 1894); С. A. Bischoff вместе с Р. Walden, "Handbuch der Stereochemie" (1894); А. Hantzsch, "Précis de Stéréochimie" (1896); H. Landolt, "Das optische Drehungsvermögen" (2 изд., 1898); Н. Вальден, "Материалы к изучению оптической изомерии" (1898: отдельными главами напечатано в т. XXX "Ж. Р. ф.-хим. общ.", стр. 483, 491, 502, 632 и 767); J. H. van't Hoff, "Vorlesungen ü. theoret. u. phys. Chemie" (2 тетр., "Die chemische Statik", стр. 94—123, 1899); P. Freundler, "La Stéréochimie" ("Scientia", № 3, 1899). Рефераты С. A. Bischoff'a в Meyer's "Jahrbuch der Chemie", отд. "Organische Chemie" — "Stereoisomerie" с 1891 г., и П. Вальден, "Оптическая изомерия" ("Ж. Р. ф.-х. о.", т. XXXII, [2], 42—59); многочисл. отд. статьи в спец. хим. журн.; E. Fischer'a в "Ber. d. deutsch. Chem. Gesellschaft".A. И. Горбов. Δ.
Энциклопедический словарь Ф.А. Брокгауза и И.А. Ефрона. — С.-Пб.: Брокгауз-Ефрон. 1890—1907.